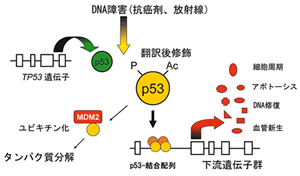
図1 |
TP53遺伝子産物p53はDNA傷害をはじめとする各種細胞ストレスによりリン酸化,アセチル化などの翻訳後修飾を受け活性化され (正の制御),複数の下流遺伝子を塩基配列特異的に転写活性化する.一方,p53はMDM2などのユビキチン・リガーゼの作用によりユブキチン化され分解される (負の制御). |
─────
総 説
─────
p53とヒトのがん
東北大学加齢医学研究所 癌化学療法研究分野
東北大学病院 腫瘍内科
石 岡 千加史
要 旨
TP53がん抑制遺伝子の変異はヒトがんの中で最も高頻度に見られる遺伝子異常である.遺伝子産物p53の機能やその周辺の分子機構を含めたp53経路の細胞生物学的な役割が次第に明らかになる一方,がん患者由来の腫瘍組織を用いた膨大な研究成果が蓄積しており,p53がん抑制タンパク質経路の破綻ががんの進展に重要な役割を担っていることが判明している.p53経路には多くの分子が関与するが,なぜp53の不活性化が重要であるのか? p53経路破綻は多くの場合TP53遺伝子内のミスセンス変異により起こるが,これはなぜであろうか? さらに,予後や治療感受性との関連についてどこまでエビデンスがでているのか? このような視点からTP53遺伝子変異のヒト「がん」における意義について,最近著者らが行っているTP53遺伝子変異の機能評価に関する研究結果交えて解説し,さらにp53研究の臨床医学へのインパクトについて海外の状況を中心に解説する.
加齢研誌56(1), 1-34, 2004.
| |
||
|
はじめに
TP53がん抑制遺伝子は393アミノ酸からなる核内タンパク質p53をコードする.p53の主な分子機能は転写活性因子であり,DNA傷害などの各種細胞ストレスやがん遺伝子シグナルなどによりリン酸化,アセチル化などの翻訳後修飾により活性化された後,複数の下流遺伝子の転写調節領域内に存在する特異的塩基配列[(5′-PuPuPuC(A/T)(T/A)GPyPyPy-3′)]×2に結合し転写を活性化する
(図1)1).p53は,塩基配列特異的DNA結合に重要なp53タンパク質の中心部第100-300残基を占めるコア・ドメイン (DNA結合ドメイン),そのN末端側とC末端側にはそれぞれ転写活性化ドメイン,4量体形成ドメインのほか,リン酸化やアセチル化残基や多くのタンパク質結合部位があり,p53の機能調節に重要な部位と考えられている2−9).これまでに多くのヒトがんにおいてTP53変異が報告され,腫瘍の種類によるがヒトがん全体の約50%
にTP53変異があり10,11),また,遺伝子増幅によるMDM2過剰発現12,13) やヒト・パピローマウイルス感染によるHPV
E6発現により14),p53のユビキチン化とプロテアソームでの分解15−17) によりp53機能が破綻する腫瘍も存在する.このため発癌やその進展過程に最も重要な遺伝子であると考えられている.p53は細胞周期のG1期/S期境界での停止,アポトーシス誘導,DNA修復促進や血管新生抑制などの重要な細胞機能に関わっている3−9).それぞれの機能を担うと考えられる下流遺伝子がこれまでに複数同定されている18,19) が,p53の活性化により,各機能がどのような機序で調節されているか十分に解明されていない.一部の遺伝子は特異的リン酸化部位のリン酸化により選択的に転写活性化されるとの報告20) や,DNA結合ドメインに結合して下流遺伝子の転写活性化を選択的に調節する機構21) が報告されている.従って,転写後修飾を受ける残基やタンパク質間結合に関与する残基のアミノ酸置換が特定の下流遺伝子の転写活性化に影響を及ぼす可能性がある.最近では,転写非依存性のアポトーシスの存在が明らかにされ注目されているが22),転写を介したアポトーシスとの間にどのような制御機構により使い分けられているのか不明である.このようにp53経路には活性化,翻訳後修飾,下流遺伝子など多くの遺伝子,遺伝子産物の関連性が指摘されているが,それではなぜp53変異は高頻度に見つかるのであろうか? この疑問に対する完全な回答はまだ得られていない.p53とヒトのがんの関わりについて理解するために,本総説の前半はp53の機能とヒトがんとの関連について解説する.ただし,この部分のより詳細な情報を必要とする場合は他の総説を参照されたい2−9).中盤には,なぜTP53遺伝子変異の多くがミスセンス変異であるかについて我々の最近の研究とTP53変異データベースからの情報を交えて概説する.なお,TP53変異データベースに関しては,Soussiらの総説11,23) も参照されたい.後半には,p53と予後,抗がん剤感受性,さらにはp53に関連するあたらしい分子標的治療法の開発の現況について解説する.なお,抗がん剤・放射線感受性とp53に関わる部分は,
El-Deiryらの総説24) も参照されたい.
 |
||
|
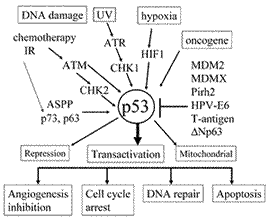
DNA傷害シグナル伝達とp53の活性化機構
p53はDNA傷害,低酸素などの細胞ストレスやがん遺伝子のなどにより活性化される (図2)8,9).この中でDNA傷害時のセンサーシステムには多数のタンパク質が関与することが判明している.様々な種類のDNA傷害のうち電離放射線によるDNA鎖切断に対してはクロマチン構造変化をきっかけにATM
(ataxia telangiectasia mutated) が活性化される25).また,紫外線照射によるDNA傷害の場合は,DNA複製の中断や,転写の中断によりATR
(ataxia telangiectasia and Rad3 related kinase) が活性化される26).ATM,
ATRや他のkinaseはp53を含めた共通のタンパク質やそれぞれの固有のタンパク質をリン酸化して活性化する27,28).このような基質タンパク質にはATMにより活性化されるCHK2,
MDM2, 53BP1, ABL, BLM, NBS1, RAD9, FANCD2, ATRにより活性化されるCHK1, HUS1, 両者により活性化されるBRCA1,
RAD17, PLK, E2F1などが含まれる.これらのリン酸化の基質タンパク質にはDNA修復,細胞周期調節やアポトーシス誘導に関わる遺伝子産物が多く含まれる.その中でp53の活性化にはATM,
ATRによるSer15のリン酸化27,28) とCHK2によるSer20のリン酸化29−31) により,p53-MDM2結合が抑制されるとともに核外輸送障害により核内にp53が蓄積すること重要であると考えられる.最近,p53の核内蓄積にはp53-MDM2結合非依存的機序があることを示唆する報告32) や,MDM2の発現量によるp53のユビキチン化の違いにより,p53分解か核外輸送かが決定されるとの報告があり33),p53の活性化機構の最終段階である核内蓄積に関しても解明すべき点は多い.また,アポトーシス誘導に関するp53の核外
(細胞質) 機能については,p53活性化以降の細胞内局在調節機構が関与すると考えられるが,不明な点が多い.
‘guardian of the genome' としてのp53
DNA傷害によって活性化されたp53は細胞をG1/S移行期に停止し,ゲノム上のDNA傷害修復のために猶予を与え,DNA傷害が生じた細胞がS期に移行しないように監視する
‘guardian of the genome' と考えられている34).このG1/S移行期に停止にはp21WAF1遺伝子の転写活性化35) が最も重要である.しかしながら,‘guardian
of the genome' としてのp53はG1/S移行期以外にG2/M期も含めた細胞周期チェックポイント機構を有するほか,DNA修復,老化,血管新生,アポトーシス誘導など,細胞の様々な機能に関与している8,9).そしてそれぞれの細胞機能に対応したp53の下流遺伝子が同定されている18,19).これらの細胞機能の一つが障害されることは発がんに有利に作用すると容易に考えられるが,多くのがんにTP53遺伝子に変異が認められることは,これらの機能が同時に障害されることが発がんに重要であることを意味する可能性がある.細胞周期チェックポイント機構は細胞老化に重要な機能であり,またアポトーシス誘導機構とともに細胞に備わった発がん防止の安全機構でもある.このため細胞周期チェックポイント機構の破綻はがんの必要条件である.さらに,ゲノム上に生じた異常に対する安全機構にDNA修復機構がある.細胞増殖制御の破綻のみならず,これらの安全装置の破綻が発癌に重要であることは,遺伝性腫瘍の原因遺伝子の機能から考えても明らかである.例えば,増殖因子受容体であるRET,
METならびにKIT受容体型チロシンキナーゼの活性型変異により,それぞれ多発性内分泌腫瘍症36),遺伝性乳頭状腎細胞癌37) および遺伝性GIST38) が発症し,細胞周期制御のRB1,
DNAミスマッチ修復遺伝子群ならびにアポトーシス,細胞周期制御,DNA修復の安全機構に関わるTP53やCHK2の機能喪失変異により,それぞれ網膜芽細胞腫39),遺伝性非ポリポーシス大腸癌40−43),Li-Fraumeni症候群44,45) が発症するなどである.
アポトーシスとがん
細胞に備わった安全機構には細胞周期チェックポイント機構以外にアポトーシス誘導がある.アポトーシスは個体の発生,老化,がん抑制に重要な役割を担う.アポトーシスは複数のタンパク質が複数の経路で関与するが,その一連の過程を3段階に分けると,1)
アポトーシスの始動に必要な外的,内的刺激とその細胞内伝達,2) BCL2ファミリーによるアポトーシスシグナルのミトコンドリアへの伝達とミトコンドリア外膜透過性制御,ミトコンドリアからのチトクロームcなどの放出,3)
システイン・プロテアーゼであるカスパーゼの活性化とDNaseによるDNA断片化 (不可逆的反応),である.1980年代,がん遺伝子の活性化による細胞増殖シグナルに関する研究が盛んになり,発癌の重要な分子機構が次々に明らかにされた.一方,B細胞性リンパ腫におけるt(14
; 18):(q32 ; q21) 転座によるBCL2の過剰発現と,そのために引き起こされるアポトーシス抑制46) など,アポトーシスとがんの関連性が指摘されるようになった.その後,多くのがん遺伝子の活性化は細胞増殖を正に制御するだけでなくアポトーシスを活性化することも明らかにされた.がん遺伝子シグナルがアポトーシスを誘導する制御機構は十分に解明されていないが,この一見矛盾するような事実は,がん化にはがん遺伝子の活性化による細胞増殖促進に加えて,アポトーシスによる細胞死を抑制する必要があることを意味している.発がん過程で様々ながん遺伝子やがん抑制遺伝子の変異が生じるが,その多くは細胞増殖シグナルの活性化とアポトーシスの破綻を引き起こす.このため,細胞のがん化を防ぐ安全機構の一つであるアポトーシスの破綻はがんのもう1つの必要条件であると言える.
 |
||
|
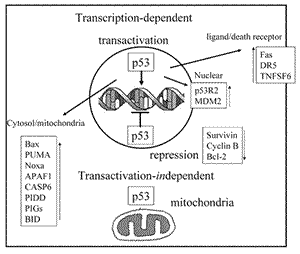
アポトーシスとp53
p53機能の中でアポトーシスは最も盛んに研究されている機能である.これはp53依存性アポトーシスに関する分子経路が,がん治療の新しい分子標的となる可能性があるからであろうか.がん抑制におけるp53のアポトーシス機能の重要性は,野生型と一部の変異p53を用いた発現系において,抗がん剤や放射線照射によって活性化されたp53が転写標的遺伝子の転写調節領域に結合する能力がアポトーシス誘導と相関することが大きな根拠になっている2−5).アポトーシス経路の各段階
(図3) に多くのp53の標的遺伝子が関与している.外来性経路では,Fas/CD9547), DR548) などのdeath
receptor, FasリガンドのTNFSF649) が転写活性化される.また,BCL2ファミリーによるアポトーシスシグナルのミトコンドリアへの伝達に関しては,BAX50), PUMA51), Noxa52), BID53) が転写活性化される.さらに,アポトーシスの下流の共通機構にではAPAF154)
やcaspase 6遺伝子のCASP655) が転写活性化される.このようにp53はアポトーシスの主要な経路を担う遺伝子を転写活性化するが,p53によって転写活性化される遺伝子として発見されその遺伝子産物がアポトーシス誘導に関与するものとしてPERP56), PIDD (p53-induced
protein with a death domain)57), p53DINP1 (p53-dependent
damage-inducible nuclear protein 1)58), p53AIP1 (p53 apoptosis-inducing
protein 1)20) などがある.p53DINP1はキナーゼ複合体と結合してp53のSer46をリン酸化し,p53AIP1の特異的な転写活性化を促進すると考えられている58).アポトーシス経路とは別に,細胞生存シグナル伝達系
(抗アポトーシス系路) にPI3-Akt経路があるが,p53はPI3-Akt経路の負の調節因子であるがん抑制遺伝子PTENを転写活性化する59) ことによりPI3-Akt経路を抑制し細胞のアポトーシス感受性を高めるものと考えられる.一方,p53には転写抑制機能があることが知られているが60),p53によるアポトーシス誘導と転写抑制の関連性が報告されている61).このようなp53により転写抑制される遺伝子の1つBIRC5遺伝子はSurvivin
(IAPの一種でアポトーシスを抑制する機能のほか,最近では細胞分裂に関与することが判明) をコードし,多くの腫瘍で発現が亢進していることからがん抑制との関連で注目されている62).p53による転写抑制は,低酸素などのある条件下においては転写活性化よりもアポトーシス誘導に重要な役割を果たしている可能性がある63).さらにp53のアポトーシス誘導能には転写非依存性機構の重要性が指摘されている64−69).特に細胞ストレス後,チトクロームc放出や下流のcaspase活性化に先立ってp53がミトコンドリアに局在すること70),ミトコンドリアに局在性とアポトーシス誘導能にTP53多型による違いが見られることなど71),興味深い研究結果が報告されている.現時点では,転写依存性機構と転写非依存性のどちらが重要であるか,さらに2つの機構の使い分けの機構があるかについては不明である.p53はアポトーシス誘導以外にも細胞周期チェックポイント機構,DNA修復,血管新生など多様な機能に関わっているが,p53依存性アポトーシスの制御はどのように行われているのであろうか? p53の機能発現には主にp53の安定化による量的調節機構が働く.既述のようにDNA傷害による特定残基のリン酸化によりMDM2の結合抑制が生じ,p53の分解が抑制される.p53が安定化すると下流遺伝子の転写活性化が起こるが,この場合,発現量によって下流遺伝子の転写調節領域への結合に関するあ閾値が異なると考えられる72).従って発現量の多少により転写される遺伝子を制御している可能性がある.もう一つの可能性は,細胞ストレスの種類によってp53の翻訳後修飾が異なる可能性である.ガンマ線と紫外線では転写活性化される遺伝子が異なるとの報告73) や,DNA傷害と低酸素では翻訳後修飾が異なることが報告されている63).Odaら20) は,Ser46のリン酸化がアポトーシスに重要で,この場合アポトーシス誘導に関わるp53AIP1遺伝子の転写活性化が特異的に転写活性化されると報告している.しかしながら,マウスではSer46は保存されておらず,リン酸化以外にも特定リジン残基のアセチル化,SUMO-1によるsumolationやPIN1によるisomerizationなどの翻訳後修飾が下流遺伝子の転写活性調節に関与している可能性もある74−79).今後, p53の機能発現に関わる制御機構の解明が必要である.
p53変異とヒトのがん
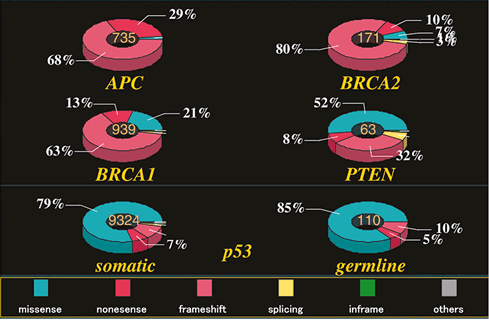
膨大なp53関連研究 (PubMedでキーワード「p53」で32,833論文が検索される) により,p53タンパク質の機能とがんの発生進展への関与が明らかにされている
(PubMed検索でキーワード「p53」AND「cancer」で23,784論文).また,1989年に最初のp53変異が報告されて以来80),14年間にTP53変異は世界中で多種類の腫瘍型について多数の検体で調べられ
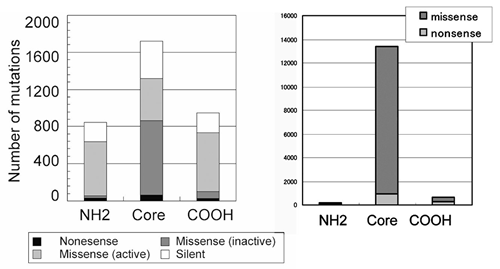
(PubMed検索でキーワード「p53」AND「mutation」で11,523論文),フランスの2大p53変異データベースに情報が蓄積され81,82),分子疫学的解析に用いられてきた.2004年現在,データベースに登録された変異は19,000件に昇り,約75%
はミスセンス変異である.これは,ヒトがんに見つかった他のがん抑制遺伝子変異の大部分がナンセンス変異・またはフレームシフト変異であることと比較すると際立った特徴になっている
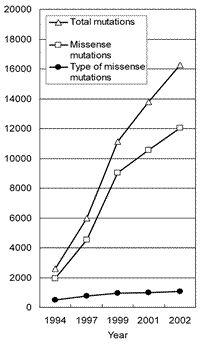
(図4).このように蓄積された研究から,いくかの基本的な疑問が生じる.全てのTP53変異はp53の機能を障害しているのか? 変異はなぜDNA結合ドメインに集中し,N末端の転写活性化ドメインやC末端のオリゴマー形成ドメインに少ないか? ミスセンス変異はなぜ多いか? 低頻度の変異は病的な意義があるか? 等々未だに不明な点も多い.一方,データベースへの登録変異数は年々増加するがミスセンス変異の種類は約1,000種類と頭打ちになっている83) ことから,発癌に関わる病的変異はほぼ出揃ったと考えられる
(図5).従ってTP53変異データベースはTP53遺伝子変異を通じたヒトがんの分子疫学的研究84−86) に有用である.さらに,p53のタンパク質構造については,DNA結合ドメインやC末端のオリゴマー形成ドメイン構造がX線結晶構造解析やNMR解析で報告されている87−89).このため,TP53変異データベースはp53変異とタンパク質構造の関連性やp53の構造と機能の関連性を知る上で貴重な情報源であると考えられる81,82).一部のTP53変異や既知のp53機能や構造の情報から「TP53変異による1塩基置換によりp53構造が変化し,p53は塩基配列特異的DNA結合能を失い,下流遺伝子を活性化できないため,細胞内での機能を発揮できない」という単純かつ明快でp53研究の基本となる学説が導かれている.しかしながら,変異の種類によって機能的意味での個性があることが一部の変異について報告されている.変異p53がp53機能や構造に直接どのような影響を及ぼすかについては全て調べられたわけではない.逆に言えば,個々の変異がp53機能や構造に直接どのような影響を調べることにより,p53の変異とがんに関わる機能
(がん抑制と抗がん剤感受性) や,構造との関係をより深く知ることが可能になると思われる.
 |
||
|
 |
||
|
網羅的変異p53ライブラリーの作成
そこで,p53とがんの関係を説明する基本的な学説に対する疑問について再評価するために,我々の研究室では網羅的ミスセンス変異ライブラリー作成し,転写因子としての機能を出芽酵母での転写機能喪失を指標にTP53変異の意義について再評価を試みた.まず,1塩基置換によって生じうる全ての1アミノ酸残基
(2,314種類) をp53 cDNAに構築し,発現変異p53タンパク質の塩基配列特異的転写活性化能を出芽酵母で評価した83).変異p53ライブラリーの作成に先立って,p53の第2コドンから第393コドンに1塩基置換をデザインした.各コドンに9種類の1塩基置換が可能であるが,このうちアミノ酸置換を伴わないサイレント変異とナンセンス変異は除外した.さらに,2種類の塩基置換が同一アミノ酸置換を生じる場合は,報告変異数の多いもの,または変異が報告されていない場合はヒトの遺伝子で出現頻度が高いコドンを優先した.このようなルールをもとに部位特異的1塩基置換導入のための合成DNAオリゴヌクレオチド2,314種類を合成した.合成オリゴは26塩基からなり,5′
側から14番目の塩基に上記のミスマッチ塩基を導入した.これらの合成オリゴを使用したPCR-メガ・プライマー法によりTP53cDNA上に1塩基置換を有するcDNAを合成し,出芽酵母のgap
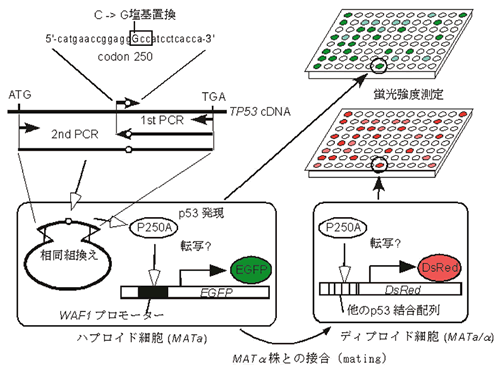
repairを利用したいわゆるin vivoクローニング法にて酵母内で各変異p53タンパク質を発現するクローンを2,314種類作成した.図6に部位特異的変異導入法による2,314種類のp53ミスセンス変異作成過程の概略を示す.合成オリゴから酵母クローンの作成まで原則的に96ウェル・フォーマットで行った.この変異p53ライブラリーは1塩基置換によって生じうる全ての1アミノ酸置換型変異p53を網羅し,これまで報告された1アミノ酸置換型変異の95%
以上を含む (一部の腫瘍では2塩基置換も報告されているが,それらは含まない).
p53ミスセンス変異体の塩基配列特異的転写活性化能
変異ライブラリー作成に使用した酵母細胞にはあらかじめp53結合部位を含むp21WAF1遺伝子のプロモーターを挿入したGFPリポータープラスミドが導入してある83).これは多数の変異体の機能を定量的に評価するために,栄養要求性レポーター遺伝子を用いた従来の酵母発現系90) を改良したものである.このアッセイ系により1アミノ酸置換を有する2,314種類の変異p53について塩基配列特異的転写活性化能をGFPの螢光強度を指標に定量的に測定することが可能である.さらに,他の7種類のプロモーター由来p53結合配列を挿入したDs-Redリポータープラスミドを保持した別の酵母株との接合
(mating) を行った.2,314種類の変異p53について8種類のp53結合配列に関する転写活性能を調べた結果 (図7),明らかに,コア・ドメイン内にある変異p53の転写活性化能はN末端側とC末端側の変異p53よりも低く,第96残基と第286残基間が機能解析によって決定されたDNA結合ドメインである.これは,タンパク質分解による構造的安定性から決定されたコア・ドメインとよく一致する91,92).この機能解析の結果に,解析対象としなかった1塩基置換
(機能に影響がないサイレント変異と機能を失うナンセンス変異) を加えた約3,000種類の「1塩基置換モデル」83) を作成した
(図8).これによると,DNA結合ドメインの1塩基置換はN末端側とC末端側の1塩基置換よりも感受性が高いことは明らかである.変異データベースに報告されている1塩基置換と比較すると,1塩基置換モデルのほうがN末端側とC末端側の機能消失変異がやや高いが,これは多くの論文における腫瘍のTP53変異解析がコア・ドメインに限定されるため,データベース上N末端側とC末端側変異の報告が少ないのであろう.逆にこのことはp53変異解析には上N末端側とC末端側の変異検索の必要性を意味する.
 |
||
|
 |
||
|
p53の機能・構造・変異相関
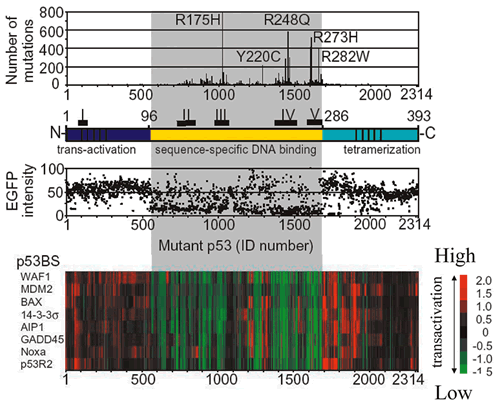
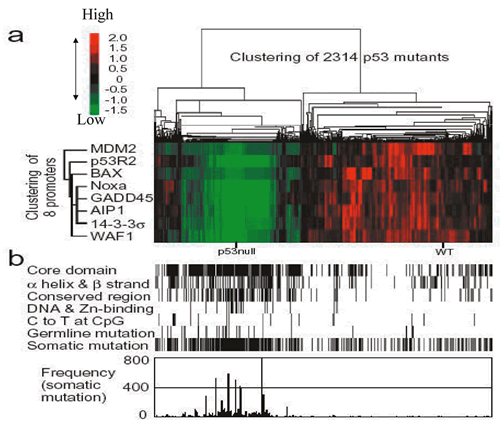
2,314種類の変異に野生型とp53を発現しない酵母をコントロールに加えた合計2,316種類のクローンについて合計8種類の異なったp53結合配列を介した転写活性化能を定量的に評価し,変異とプロモーターに関する階層的クラスター解析を行った
(図9).その結果,各変異p53は,野生型p53を含み転写活性能を有するクラスター (全体の約3分の1) と,p53を発現しないネガティブコントロールを含み転写活性が障害されているクラスター
(全体の約3分の2) に大別された.この変異p53の機能によるクラスター化の結果を,構造に関する各種指標 (p53のドメイン,αへリックス・βストランドの2次構造,DNAまたは亜鉛結合残基)
と,前述のデータベース上に報告された変異と対比した.図10に示すように,機能障害を有する変異p53はDNA結合ドメインであるコア領域 (コドン100から300)
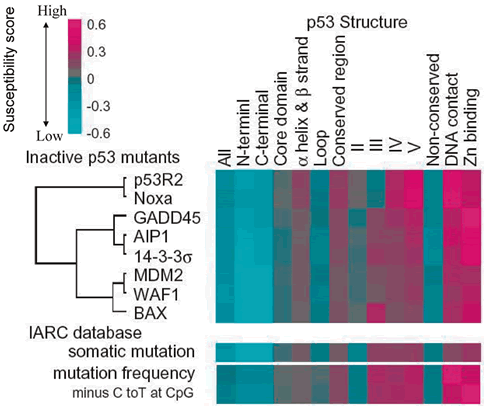
に集中していることが明らかになった.さらに機能障害を有する変異p53は,αへリックス・βストランドの2次構造に集中し,2次構造を連結するループ構造には少ない傾向があること,DNAまたは亜鉛結合残基上の変異の大部分は機能障害を有することが明らかになった.すなわち,p53の機能と構造には明らかな関連が認められた.各変異の機能とTP53変異の報告を比較すると,体細胞性変異および生殖細胞系変異は明らかに機能障害クラスターに集中していた.また,体細胞性変異の報告頻度との関係も同様であった.機能・構造相関と同様に,機能・変異相関も明らかに認められた.図11に各変異p53の機能,変異前のアミノ酸残基の保存性
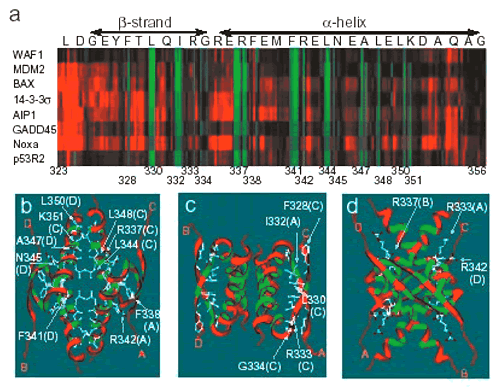
(構造の指標),および対応する変異頻度を示した.3者 (機能,構造,変異) 間には明らかな相関性が認められた.前述のようにC末端側の報告変異は少ないが,この中で4量体形成ドメインの一部の残基は変異に対して感受性が高い.実際,この残基内には腫瘍由来のミスセンス変異やLi-Fraumeni症候群の変異が報告されている93,94).4量体形成ドメインのX線結晶構造解析の結果と機能について関連を調べた
(図12).その結果,2量体形成や4量体形成に必要と予測されている残基 (330, 332, 337, 341, 344) は確かに変異に対して感受性が高く,特にαへリックス間の疎水性残基
(分子間で直接接する残基) は感受性が高い.我々の研究室では個々の変異の4量体形成と転写活性化能との関係も調べ,4量体形成がp53の転写活性に重要であることを明らかにしている
(加藤,川口ら,未発表).
 |
||
|
 |
||
|
 |
||
|
 |
||
|
 |
||
|
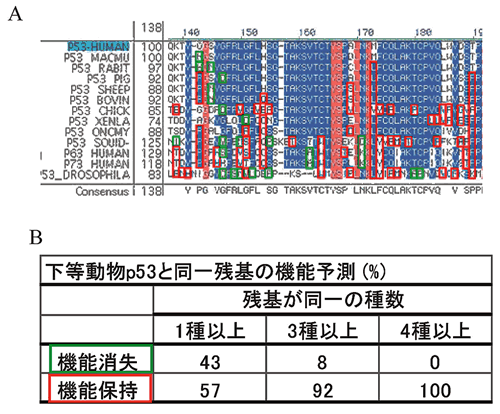
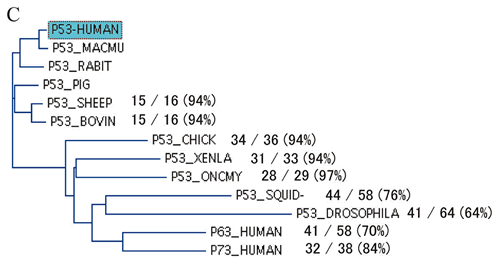
アミノ酸残基の保存性からみた機能
p53のDNA結合ドメインのアミノ酸残基は下等動物から種を越えて良く保存されており95),アミノ酸残基保存率を指標にした解析では,機能と変異ともよく対応することは前述のとおりである.そこで今度は,ヒトp53とは一致しない下等動物のアミノ酸残基を指標に変異p53の機能的保存性を検討した.まず,p53ホモログ・オルソログ間でアミノ酸残基のアライメントを取り,下等動物のアミノ酸残基でヒトp53と一致しないものを拾い上げ,我々が作成した変異p53とアミノ酸残基が一致するものを抽出した.我々の研究では,1塩基置換を条件にミスセンス変異を作成しているため,下等動物でヒトと異なるアミノ酸残基の一部しか合成していないので,このようない下等動物のアミノ酸残基はコドン100から290の範囲に作成したミスセンス変異1,029変異の9.7%,100種類あった.そこでこれら100種類について対応するヒトでのミスセンス変異の機能を「機能消失」と「機能保持」としてスコアをつけた.この場合,野生型よりも機能は弱いが機能を部分的に保持している場合は「機能保持」と分類した.その結果
(図13),100残基のうち57% は「機能保持」,43% は「機能消失」であった.この領域に作成した1,029種類の変異は「機能消失」が538種類
(52%),「機能保持」が491種類 (48%) であり (P=0.076,カイ2乗検定) ,下等動物に出現する残基という情報から機能を予測することの信頼性はさほど高くないと考えられる.しかしながら,下等動物間で変異残基が何種類の種に共通して見られるかという情報は有用で,3種類以上の種で見られる変異残基は
「機能消失」は3種類 (8%),「機能保持」は36種離 (92%) (P<0.001,カイ2乗検定),4種類以上の種で見られる残基は,100%「機能保持」であった.また,ヒトp53と不一致残基の機能保存性を下等動物ごとに調べると,哺乳動物のみならず脊椎動物で高い機能保存性が認められた.この手法は,p53以外のタンパク質に関して,そのcSNPが機能に及ぼす影響を下等動物のホモログ・オルソログとの比較で予測できる可能性を示し,とくに機能が不明なタンパク質の場合有用であると考えられる.
  |
||
|
 |
||
|
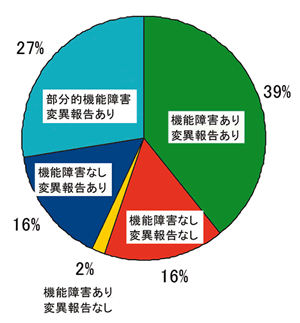
機能・変異仮説と個々の変異
以上のように,我々の研究ではTP53変異の結果生じる変異p53の機能障害とタンパク質構造間には総じて強い関連性があることを示したが,この結果は発癌に関わるp53の役割を考えた場合予想どおりの結果である.しかしながら,個々のTP53変異に関しては多数の例外が見られることが明らかになった.機能と変異についの解析では,54.7%
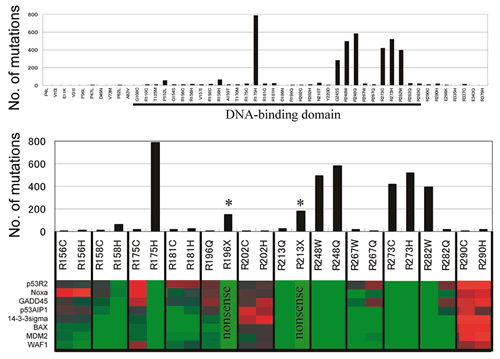
の変異 (1,266種類) は「機能・変異仮説 (機能障害を伴う変異がクローン選択の結果として腫瘍に見つかる)」で説明可能であった (図14).すなわち905種類
(39%) は全てのp53結合配列に関して機能を有しており変異の報告は認められていない.また,361種類 (16%) の変異は機能障害があり変異も報告されているもので,R175H,
R248Q, R273Hなどの高頻度変異はこのタイプである.残りは機能・変異仮説では簡単に説明できない変異であり,この群はさらに3タイプに分類される.第1は,373種類
(16%) の変異で,p53結合配列に関して機能を有しているにも関わらず変異が少なくとも1回は報告されているものである.このタイプの変異は機能的には野生型と同じで病的意義はないが,クローン選択の段階でそのTP53変異アレルを持った細胞が偶然に選択されてきた可能性が考えられる.実際,この種類の変異が報告されているがんでは,データベース上にTP53変異が複数存在する場合がある.このほかにも,このタイプの変異は部分的にp53の機能が障害されており,弱い病的意義を意味している可能性も否定できない.第2は39種類
(2%) の変異で,これらは病的意義があるが遺伝子上の変異頻度が低いか,または多くがDNA結合ドメイン外にあることから,変異の検索が十分に行われていない可能性が考えられる.
 |
||
|
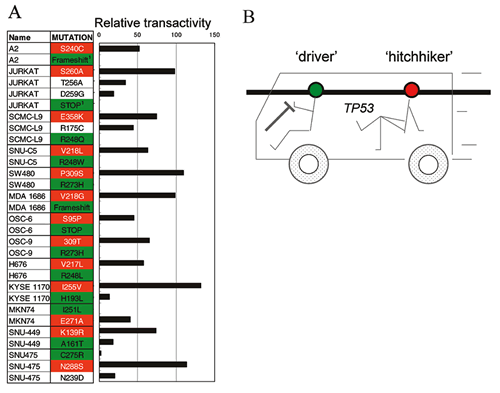
転写活性の部分的機能障害
第3は,635種類 (27%) の変異で,このタイプは一部のp53結合配列に対する機能が障害されている.我々はこのタイプの変異グループを多様性p53変異体と呼んでいる.多様性p53変異体の報告回数は機能障害のあるp53結合配列数に依存することから
(図15),発がんにおける病的意義が高頻度変異と野生型の中間的である可能性が考えられる.ミスセンス変異の種類によって複数の変異p53が部分的な機能障害になる可能性は,最近,Resnickらによっても明らかにされている72).このようなミスセンス変異に関する機能的多様性は,p53経路に対する異なった影響をもたらす可能性を示唆し,今後,変異と予後,抗がん剤感受性との関連を見直すことは非常に興味深い.そもそもp53経路の破綻に関しては,TP53遺伝子変異とp53経路のほかの遺伝子変異との違い,TP53遺伝子変異のなかでも欠失変異とミスセンス変異の違い,さらには,マウスモデル96) やLi-Fraumeni症候群97,98) におけるTP53欠失変異とミスセンス変異の種類による腫瘍ペクトラムの違い,H175P,
R181Pなどの一部のミスセンス変異がアポトーシスや細胞周期停止に関する機能が高頻度変異と異なること,などが知られている.このような機能的多様性は,p53経路の破綻箇所やp53変異の種類によってがん抑制に関する細胞生物学的な影響が異なり,予後や抗がん剤感受性に影響を及ぼすことは想像に難くない.また,このようなミスセンス変異の機能的意味での多様性から,我々は,温度感受性変異99),アポトーシス誘導能が野生型p53よりも強力な
‘super p53' (角道ら,未発表),腫瘍由来の高頻度変異の機能を回復させる第2の点変異 ‘2nd-site suppressor' (大塚ら,未発表)
を多数同定している.今後,このような研究ががんの個性を明らかにするための診断や新しいがんの分子標的治療法の開発につながる可能性があり注目している.
ドミナント・ネガティブ変異と機能獲得変異
以上,塩基配列特異的転写活性可能を指標にした機能喪失モデルから変異p53の機能に関する多様性について示したが,ミスセンス変異によっては野生型p53の機能を抑制するドミナント・ネガティブ変異や新たな機能を獲得している変異の存在が以前から知られている100−102).これは一部でミスセンス変異を有するTP53アレルは存在するがLOHが見られず野生型アレルが残存している腫瘍が見られること,既述のようにミスセンス変異型のLi-Fraumeni症候群やTrp53ミスセンス変異マウスの表現型がその他のタイプの変異と異なること,などが理由であるが,実際に野生型p53と共発現して転写活性化能に関するドミナント・ネガティブ効果が調べられた変異の種類はそれほど多くはない103).我々は変異ライブラリーを用いて野生型p53の転写活性に対するドミナント・ネガティブ変異を網羅的に調べてDNA結合ドメインに多くのドミナント・ネガティブ変異を同定している
(未発表).この他に変異によっては野生型p53に依存しないドミナントな機能獲得型変異がある104,105).
 |
||
|
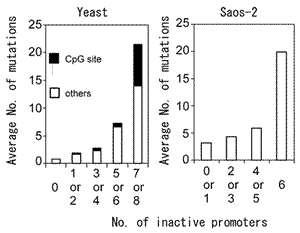
頻度の低い変異の解釈
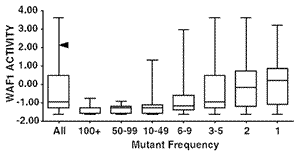
先の述べたTP53変異データベースのうちミスセンス変異は75% で約1,000種類ある.この中には250回以上報告されているいわゆるホットスポット変異
(R175H, G245S, R248Q, R248W, R249S, R273C, R273HおよびR282H) から1回しか報告されていないものまで含まれる.2,314種類の変異p53の転写活性化能に関する情報をもとに,データベース内の約1,000種類のミスセンス変異について再評価を試みた83,106).その結果,報告頻度が高い変異の大部分は明らかに機能喪失変異であったが,報告頻度が少ない
(5回以下) の変異の50% は機能を部分的または野生型p53同様に保持していることが判明した (図16).これは腫瘍の種類によらず共通に認められた106).このことは低頻度に報告された変異は直接病的意義がないものが含まれている可能性を示唆する.この点に関して,T.
Soussiは我々との共同研究でTP53変異報告論文のメタアナリシスにより,野生型p53同様に保持している低頻度のミスセンス変異が特定の論文に集中して見つかっていることが判明し,そのような変異は検出方法に問題があるエラーであることが予測されている.また,変異の表記の際に生じたエラーが相当数含まれている可能性があることも同時に指摘している
(T. Soussi,私信).野生型p53同様の機能を保持している低頻度のミスセンス変異の解釈にはもう一つの可能性がある.ゲノムDNAのCpG 2塩基配列部分は,C:G to T:Aトランジションの好発部位であり,TP53遺伝子変異の場合でも全点変異数の約30%
が,このタイプの変異である.これはCpGのcytosineのメチル化による5-メチルシトシンの生成と,5-メチルシトシンの脱アミノ化の結果生じるT/Gミスマッチがシトシンの脱アミノ化の結果生じるU/Gミスマッチよりも修復効率が悪いための変異であると考えられる.TP53遺伝子エクソン5-8内に存在するCpG部位の46
cytosine塩基は,ヒトの正常組織では組織特異性なく全てメチル化されていることが確認されている107).この他にも,メチル化シトシンは内因性変異原や紫外線,Benzo(a)pyrene
diol epoxide (BPDE) などの外来性変異原にも親和性があると言われている.図17上段にTP53cDNA内の各CpG部位ミスセンス変異のIARC変異報告頻度を示す.DNA結合ドメインのCpG変異が多く報告されていることは,変異の頻度と機能との関連性を裏付ける.しかしながら,DNA結合ドメイン内の同一コドンに生じる変異の報告頻度にはときに大きな差があることなど,不明な点がいくつかあった.我々の行った詳細な機能解析結果から,この点についていくつかの新しい知見が得られた.下段は同じコドン上のCpGにより2種類のミスセンス変異またはナンセンス変異が生じる12コドンについて8種類のp53結合配列に対する転写活性と変異報告頻度を比較したものである.同じコドンに生じたトランジションでも,転写機能を保持しているかまたは部分的に転写活性を保持しているミスセンス変異は報告頻度が低く,転写機能を完全に失っているミスセンス変異やナンセンス変異の場合は報告頻度が極めて多いことが明らかである.また,この場合,ナンセンス変異よりもミスセンス変異のほうが明らかに変異頻度が高く,この点に関しては機能喪失モデルでは説明できない.この結果はミスセンス変異には先に述べたドミナント・ネガティブ効果または機能獲得効果のような優性な効果が存在する可能性を裏付ける.ここで注意しなければならないのはCpG上のトランジションの場合,その変異が転写機能を保持していても5-20回程度の変異報告頻度があることである.このことはCpG上のトランジション変異と非CpGトランジション変異の病的意義を報告回数から単純にと比較することはできないことを意味する.R282Qの場合をはじめこのグループの変異はそれ以外のTP53変異が見つかるケースが少なからずあり,この場合いわゆるヒッチハイク型変異である可能
性が高い (図18).このような状況から逆に,R175HなどCpGトランジション変異でホットスポット変異の一部にも発癌過程で偶然ヒッチハイク型変異になったものも存在する可能性があると考えられる.
 |
||
|
 |
||
|
 |
||
|
p53変異の機能評価と分子疫学
一方, 外来性変異原のうちBPDEはDNAのグアニンN2位に付加体を形成しG to Tトランスバージョンの原因になっていると考えられ,特に肺がんにG
to Tトランスバージョンが多い原因はBPDEの直接または間接的な効果であるとされている.このBPDEの付加体形成はTP53のコドン
157, 248, 273の変異ホットスポットにも確認され,肺癌においてこれらのコドンのG to Tトランスバージョンが多いのは付加体形成の直接的な結果であるとする
‘mutagenesis' モデル108) が提唱され,特にこのモデルが喫煙と発癌を直接説明しうるモデルとして注目を集め現在でも多くの研究者に支持されている.しかしながら,その後の研究では肺癌のなかで喫煙者,非喫煙者間ではG
to Tトランスバージョンの頻度はそれぞれ30% と7% で有意に喫煙者で多いものの,変異スペクトラムは両者に差がないことから,この ‘mutagenesis'
モデルは否定的であるとする意見もある.最近では,G to Tトランスバージョンが喫煙者肺癌に多い理由は,BPDEなどの外来性変異原が細胞内代謝に変化をもたらし非DNA傷害性ストレスとして間接的に変異を引き起こし,CpG上のC
to Tトランジションと同じように,発癌過程で選択された結果であるとする ‘selection' モデルが重要視されている109).‘mutagenesis'
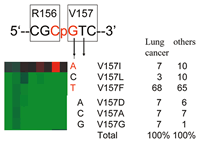
モデルを否定的にする最も特徴的なのは,コドン157の変異の解釈である.コドン157は当初BPDEのadduct形成のホットスポットと報告され,コドン157のG
to Tトランスバージョン (V157F) が同一塩基のG:C to A:Tトランジション (V157I) と比較して頻度が高いことがその説明根拠になっていた.しかしながらRodin
SN ら109) によると,肺癌以外の癌腫でも同様にG to Tトランスバージョンが多くを占めることからBPDEの直接作用は否定的である.そこで我々は該当する変異の転写機能データを当てはめたところG:C
to A:Tトランジションで生じるV157Iが機能を保持しG to TトランスバージョンによるV157Fは機能が失うことが判明した.このことはG
to Tトランスバージョンが特別に高頻度なのではなく,C to Tトランジションに病的意義がなく低頻度であるために相対的にG to Tトランスバージョンの頻度が高くなっていたと解釈される.Rodin
SNら109) は,この部位の他のミスセンス変異も機能を保持し病的意義が少ないと予測したが,我々のデータは図19に示すようにV157I以外の他のミスセンス変異はV157F同様に機能障害を伴う変異であった.これらの結果から,コドン157の変異スペクトラムは発癌過程で
‘selection' された結果を反映し,G to Tトランスバージョンは確かにコドン157に生じやすいが肺癌に非特異的であることから,BPDEの直接作用ではないことを裏付ける.
TP53変異と予後
これまでに各種がんにおいて,TP53変異または免疫組織染色によるp53発現と患者の予後についての関連について検討され,乳癌,肺癌,大腸癌,膀胱癌など複数の癌腫で予後因子としての重要性が指摘されている.ここで注意をしなければならないのはp53機能破綻の代替評価法である免疫組織染色
(IHC) の解釈である.75% のTP53変異はミスセンス変異であり核の濃染が特徴である.しかしながら,残りの25%
を占めるナンセンス変異,フレームシフト変異,スプライシング異常に関わる変異はIHCでは検出できない.使用する抗体や施設間のバラツキも少なくないと予測される.また,TP53変異を検出する方法の場合も,全翻訳領域を解析対象にしない方法やPCR-SSCP法をはじめとするスクリーニング方法は検出感度に難点がある.直接DNAやcDNAを用いて全翻訳領域をシークエンスすることが理想であるが23),この場合IHCと比べて労力が大きい.さらに,解析対象の背景,症例数,組織型,人種,病期,他の予後因子の状態などが影響するため,個々の論文のエビデンス レベルは高くない.比較的頻度の高い癌腫の場合や報告論文数が多い癌腫の場合は論文のシステマティック・レビューによる蓄積した論文の評価とメタ・アナリシスによる検証が可能であり,このうち肺癌と乳癌でp53と予後の有意な関連が報告されている.肺癌では,Mitsudomiら110) は,非小細胞肺癌 (NSCLC) 43論文を選択しIHC染色陽・陰性 (計3,579症例)
またはTP53変異の有無 (計1,031症例) についてメタ・アナリシスを行い,NSCLC全体,および腺癌の場合はIHC陽性,TP53変異有はともに5年生存率に関する負の予後因子であることを報告している.また,Huncharekら111) は,NSCLCについてTP53変異の有無と予後を調べた8論文
(計829症例) に関するメタ・アナリシスによりTP53変異有は2年生存率に関する負の予後因子であることを示している.さらにSteelsら112) は,肺癌に関する74論文
(うち30論文は単変量解析でp53は統計学的に有意な予後因子) のメタ・アナリシスを行い,臨床病期に関係なく,また使用した抗体に関係なくp53 (IHC染色陽性)
はNSCLCにおいて生存期間に関する負の予後因子であり,小細胞肺癌では十分なデータが得られなかったことを報告している.乳癌の場合は,多くの論文でp53と予後の関係が調べられている.Pharoahら113) は,16論文
(計2,319症例) のメタ・アナリシスを行い,TP53変異有がリンパ節転移の有無に関わらず全生存期間および無病生存期間に関する独立予後因子であることを示している.一方,大腸癌の場合はp53と予後の関連について肺癌,乳癌同様に多くの論文が発表されているが,これまでにメタ・アナリシスの報告はない.Iacopetta114) は,100症例以上を対象にしたp53と予後の関連を報告した25論文
(このうち20論文がIHCによる解析,5論文がTP53変異解析,3論文が両者) についてシステマティック・レビューを行い,14論文がIHC陽性またはTP53変異有が予後と負に相関,3論文は逆に正の相関する傾向を示したが,大部分の論文は統計学的に有意な解析数ではなかったと報告している.大腸癌におけるメタ・アナリシスの結果が待たれる.膀胱癌ではメタ・アナリシスの報告はないが,Schmitz-Dragerら115) らは,IHCによる解析を行った43論文
(計3,764症例) についてシステマティック・レビューを行い,単変量解析では42論文中28論文が,多変量解析では33論文中15論文で有意に予後と関連していたことを報告している.このほかにも,上部尿路系腫瘍
(腎杯,尿管)116),上皮性卵巣癌117),脳神経膠腫118) に関してシステマティック・レビューが行われているが,メタ・アナリシスによる報告はない.このようにTP53変異の有無またはIHCによる発現は予後因子となりうるが,Borrensenらのグループ119) は,p53の特定の構造に生じた変異と予後の関係について精力的に研究を続けている.彼女たちは乳癌にみつかったTP53変異のうちDNA結合ドメイン内で亜鉛結合残基を含むL2
(コドン163-195) とL3 (コドン236-251) の変異は,アドリアマイシン抵抗性と予後不良に関連することを示し,その後の複数の論文120−123) でL2/L3変異と予後との負の相関は支持されたが,Powellら124) の1,037症例による研究では否定的で,むしろエクソン4の変異と予後との負の相関が指摘されている.L2/L3変異と予後との負の相関は,NSCLC125) や食道癌126) でも報告がある.さらに,大腸癌でL3変異が予後不良127),頭頸部癌でDNAに直接接する残基の変異が予後不良128),ミスセンス変異はその他の変異に比べて予後不良129) とする報告も見られる.いずれの場合もシステマティック・レビューとメタ・アナリシスによる検証は行われていない.個々の変異の種類については解析された研究はまだ少ない.これは変異の種類が多いために個々の変異に対する症例数が少ないためである.その中で報告頻度の高い変異に関しては,大腸癌においてR175130),G245131) 残基の変異が予後不良であるとする報告がある.報告頻度の高いホットスポット変異に関しては,さらに大規模な研究を行えば予後との関連を明らかにできるかもしれない.
コドン72遺伝子多型と易罹患性,予後
コドン72の遺伝子多型には72P (コドン72がCCCでプロリンをコード) と72R (コドン72がCGCでアルギニンをコード) があり,一般人口におけるアレル頻度は高頻度で人種差が見られる
(日本人の場合,アレル頻度72R/72Pは約1.5).野生型p53の場合,72Rか72Pかにより転写活性化能には違いがないことは既に知られている.最近,我々は大部分のミスセンス変異についても72Rと72Pによる転写活性化能に違いが見られないことを観察している
(大塚ら,未発表).発がんとの関連でコドン72の遺伝子多型が最初に注目されたのは,子宮頸癌である.対照健常人との比較で,子宮頸癌患者には72R/Rホモ接合の人が多く,72R/Rが子宮頸癌の危険因子であり,その原因はヒトパピローマウイスル
(HPV) のE6タンパク質によって引き起こされるp53の分解 (p53の不活性化) の感受性が72Pよりも72Rのほうが高いことに起因するとの報告である132).しかしながら,その後72R/Rの易罹患性を指示しない論文が複数発表された.最近,Koushikら133) によるシステマティック・レビューとメタ・アナリシスによる検証が行われ,72R/Rはそれ以外
(72P/Pまたは72R/P) と比べて子宮頸癌のリスクに差がないとする結果が出ている.一方,他の癌では72P/Pが発がんリスクを高める (オッズ比が高い)
とする報告が乳癌134),大腸癌135),前立腺癌136),甲状腺癌137),移行上皮癌138),慢性骨髄性白血病139) などで報告されている.しかしながら,システマティック・レビューとメタ・アナリシスによる検証は行われていない.肺癌については13論文のメタ・アナリシスが行われ72P/Pホモ接合は72R/Rまたは72R/Pに比べて僅かに高いオッズ比
(1.18) であったと報告されている140).一方,72R/Pヘテロ接合のがん患者の腫瘍組織におけるTP53変異検索により,各種扁平上皮癌141),乳癌142),肺癌143),尿路移行上皮癌144) でTP53変異を伴った72Rアレルが高頻度に残存していることが示されている.このことはTP53変異が72Rアレル上に生じたほうが72Pアレル上に生じる場合よりも発がん過程で何らかの理由で有利であるためと考えられ興味深い.Marinら135) は,その理由として72Rアレル上に生じたTP53変異由来の変異p53タンパク質がp53ファミリーメンバーであるp73
(後述) に結合し,p73依存性のアポトーシス誘導を抑制することを報告している.また,Tadaら145) は,この72R由来の変異TP53はp53に対するドミナント・ネガティブ変異が少なく,p53に対するドミナント・ネガティブ効果がない劣性変異が多く見られることを報告している.このことは,発がん過程におけるp53経路の破綻にはp53の機能消失だけでは不十分で,72R多型またはドミナント・ネガティブ効果が必要であることを示唆し,興味深い.コドン72の遺伝子多型と予後の関係については現在まだ一定の見解が得られていない.肺癌で72P/Pの人から生じた場合予後が不良傾向であったとの報告146) や,72R/Pヘテロ接合の乳癌患者由来の乳癌組織におけるTP53が72Rである場合予後が不良であるとの報告がある147).
p53と抗がん剤感受性
アポトーシス誘導はp53の重要ながん抑制機能の一つであると考えられる.このため,抗がん剤 (ここで言う抗がん剤はいわゆる化学療法剤) や放射線治療に対する治療感受性に関与する可能性が指摘されている.抗がん剤によるストレスを受けた細胞はp53が活性化され転写依存性または非依存性のアポトーシス経路が活性化されるほか,サバイバル経路が抑制されること
(前述) ががん細胞死に貢献していると考えられる.逆にp53経路が破綻した細胞は,上記のようなアポトーシス誘導機構が正常に働かなくなり,抗がん剤感受性が低くなると考えられる.実際,腫瘍細胞株に抗がん剤を投与すると大部分の抗がん剤では変異p53を持つ細胞よりも野生型p53を持つ細胞により効果が強いことが示されている148,149).また変異p53を持つ細胞に野生型p53を導入することで抗がん剤感受性が高まることも報告されている.しかしながら,p53と抗がん剤感受性の関係は,実際の臨床研究結果からそう単純ではなくTP53変異の種類,コドン72の遺伝子多型,p53ファミリーメンバーの発現,抗がん剤の種類,正常組織の中でも組織特異性や腫瘍の種類による違いなど,複数の因子が関与しているものと考えられる.
TP53変異の種類と抗がん剤感受性
TP53変異は転写活性化能を低下させるが,報告された変異の中には野生型とほぼ同等の転写活性化能を持つ変異体が存在することが明らかになっている
(前述).さらに変異の中にはある標的遺伝子群への転写活性化能は低下するが別な標的遺伝子群については低下しない場合もある (前述).このように,報告されたTP53ミスセンス変異により発がんに関する寄与が異なる可能性があり注意を要する.報告頻度の高いものはpathogenicな変異である可能性が高いと考えられるが,比較的頻度の低いミスセンス変異の場合は,機能に基づく評価の情報が必要である
(頻度の低い変異の解釈).このことは同時に変異の種類によって細胞の抗がん剤に対する感受性に違いが生じる可能性を意味する.個々のTP53変異と抗がん剤感受性を調べた研究はないが,乳癌ではDNA結合ドメイン内の特定領域の変異
(L2, L3) が抗がん剤抵抗性119,150) であり,予後不良と関連するとの報告がある119).
p53ファミリーメンバーと抗がん剤感受性
p73とp63 (p51) はp53ファミリーメンバーである151,152).TP53のホットスポット変異と同等の変異を導入するとp63は転写活性化を失うが153),がん細胞では変異はほとんど見つからず154,155),個々のタンパク質のがん抑制に関わる貢献度はp53よりも低いと考えられる.p73とp63にはalternative
splicingによる複数のisoformがあり,転写活性化ドメインを有するTA isoformと,それを欠いたΔN isoformがある152,156−158).TA
isoformのp73とp63は強制発現実験において転写活性化する下流遺伝子にp53と共通性があり151,152,159),さらにアポトーシス誘導能を有する160).これに対してΔN
isoformは,p53の下流遺伝子の転写活性化に対してドミナント・ネガティブに作用する161−164).p53が標的遺伝子に結合してアポトーシスを起こすためにはp63とp73の存在が必要であることがKOマウスMEF細胞による研究で示されている165).興味深いことに,DNA傷害を受けた細胞においては,p53はp73またはp63が欠失してもp21やmdm2には結合できるが,Bax,
Noxa, Perpといったアポトーシス誘導性標的遺伝子には結合できないこと,逆にp63はp53が欠失した場合にp21やmdm2には結合しないが,これらのアポトーシス誘導性標的遺伝子に結合することが判明した165).p73はp53同様に抗がん剤ストレスにより誘導されp53AIP1を発現させ,細胞をアポトーシスに導く166,167).野生型p53はp63やp73とヘテロオリゴマーを形成しないのに対して,ある種の変異p53
(後述) はp73やp63と結合してその機能を阻害する168).このようにp53ファミリーメンバー間でアポトーシス誘導に関して相互に関連していると考えられる.最近,Irwinら169) は,p73が抗がん剤感受性に関連することをドミナント・ネガティブp73変異,iRNA,相同組み換えによるp73の機能破壊により示している.従って,DNA傷害後の細胞の運命はTP53変異,多型のみならずp73の発現が影響することを示唆する.
 |
||
|
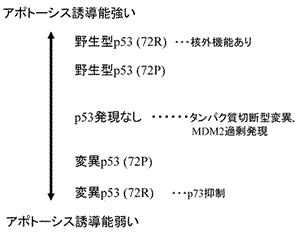
TP53遺伝子多型とp73
先に野生型p53の転写活性化能に関してコドン72の遺伝子多型72Rと72Pの間に差がないと記したが,最近,72Rは72Pよりも野生型p53のアポトーシス誘導能が強いことが示され,その理由として72R野生型p53はミトコンドリアの局在性が強く転写非依存性のアポトーシス誘導が起きやすいためと考えられている71).従ってTP53が野生型のがん細胞の場合,72R/R
(72R/Pも?) の抗がん剤感受性が高い可能性があり今後の研究課題である.また,前述のように変異p53の中にはp73と結合してp73機能によるアポトーシス誘導能を阻害するものが報告され168) 抗がん剤感受性との関連で注目されている.このような変異p53のコドン72多型は72Rである場合が多い.今後,どのような変異p53がコドン72の遺伝子多型との組み合わせでp73機能を障害し,抗がん剤感受性に影響するかを評価する必要がある.p73と変異p53の結合にはp53のコドン72の遺伝子多型によって違いがある141).すなわち,72Rの場合,72Pの場合と比べて,p73に結合しやすくかつp73によるアポトーシス誘導能を阻害しやすいことがin
vitroの系で判明している.頭頚部扁平上皮癌では,野生型TP53のコドン72が72Rである方が72Pであるよりも抗がん剤感受性が高く170),さらにTP53変異が存在する場合は72Rアレルに変異があると,72Pアレルに変異があるときよりも抗がん剤感受性が低くなる171).このように,TP53変異とコドン72多型との組み合わせで細胞の抗がん剤感受性は概ね図20のように予測される.今後の研究として,網羅的変異ライブラリー
(前述) を用いてp73機能を抑制し抗がん剤感受性を低下させるTP53変異の特定,p53に対するドミナント・ネガティブ変異との関連について明らかにする必要があろう.
その他の抗がん剤感受性因子
p53により誘導される標的遺伝子の種類は各組織によって異なることが示されている172,173).また,組織によりアポトーシス誘導に関わるp53標的遺伝子数や種類が異なる可能性がある.さらに,細胞の種類によって細胞ストレス時に細胞周期停止を起こすかアポトーシスを起こすか異なると考えられる24).加えて各組織においてストレスに対する細胞応答経路の種類やp53依存度も異なると考えられる.p53によって細胞周期が停止する細胞よりもアポトーシスに至る細胞の方が抗がん剤による感受性が高く,また,ストレス応答経路のp53依存度が高いものほどTP53変異によって抗がん剤感受性も阻害されやすいと考えられる.しかしながら,p53に対する組織特異性や反応のメカニズムはほとんど解明されていない.一般的にTP53変異は多くの抗がん剤 (アルキル化剤,プラチナ誘導体,フッ化ピリミジン系代謝拮抗剤,Topoisomerase
IおよびII阻害剤) に対する感受性を低下させると考えられるが,vincristine, vinblastine, paclitaxelのような微小管重合または脱重合阻害作用を有する抗がん剤ではTP53変異の有無で感受性が変わらないとの報告がある148).しかしながら,腫瘍細胞にpaclitaxelを加えたところp73の発現が誘導され,細胞にアポトーシスが起こり,かつp53の変異により抗がん剤感受性が左右されたとの報告もある151).一方,腫瘍細胞にアデノウイルスを用いて野生型p53を発現させた後にpaclitaxelを添加するとpaclitaxelの効果が弱くなり,理由として先に発現したp53により細胞停止が起こり,細胞分裂特異的なこの薬剤の効果が減弱した可能性があるとの報告がある174).米国NCIの60種類の培養細胞からなるがん細胞パネルを用いた研究でもpaclitaxelはより変異p53細胞により効果があるとの報告がある149).Paclitaxelに対する感受性については,他の抗がん剤と異なり変異p53やコドン72多型以外の因子が関与している可能性が高く,その分子機構の解明が必要である.マウスの系ではpaclitaxelがマクロファージからのTNF-αを放出しp53非依存性のアポトーシスを引き起こすとの報告がある175).
p53と新しいがん治療
以上のように,p53はがん抑制に重要な役割を担うと同時に,患者の予後や治療感受性にも関わっている.多くの腫瘍においてp53経路が破綻しているこおから,p53とその経路を利用した新しい治療法が開発されつつある.現在,p53を指標にした治療はまだ実用的なレベルには至っていないが,ウイルスを用いた治療法は臨床試験で安全性が確認され抗がん剤との併用療法による臨床試験での結果が待たれる.さらに,p53とその経路に特異的に作用する小分子化合物の開発が期待されており,これまでに発表された小分子化合物の概要について以下に記す.
p53遺伝子治療
多くのヒトがん細胞ではp53経路が障害されており,p53経路の再活性化はがん治療に有用であると考えられる.多くの培養細胞を用いた実験でp53の強制発現により細胞増殖抑制とアポトーシス誘導が観察されている.このような背景から初めてのがん抑制遺伝子を用いた遺伝子治療としてp53遺伝子治療が開発された.アデノウイルス・ベクターに組み込んだ野生型p53
cDNA, INGN201 (商標ADVEXIN, Introgen) を腫瘍局所に注入することによりがん細胞に強制発現させアポトーシスを誘導する方法であり,米国では脳腫瘍176),膀胱癌177),非小細胞肺癌178,179) などで臨床第I相試験が終了している.また,転移のない非小細胞肺癌で手術や放射線化学療法の適応がなかった症例に対する臨床第II相試験では,放射線照射
(60Gy) とINGN201 (3回) の外来治療を行い,CR1例を含む63% の奏効率,SDは16% を認めた180).臨床第I,
II相試験が終了した頭頸部扁平上皮癌181) などで複数の臨床第III相試験が開始されているほか,抗がん剤との併用による臨床試験も行われている182).本邦では,肺癌に対するアデノウイルスp53遺伝子治療が当研究所の西條,貫和らを含めて多施設共同研究で実施されたほか183),千葉大学で食道癌に対して行われた184).p53の発現が注射部位とその周辺に限局し腫瘍全体に広がらないこと,効果に持続性がないことなど改良すべき点も多い.
E1B欠損アデノウイルス
E1B変異によるE1B-55Kを発現しない変異アデノウイルスdl1520株 (ONYX015) による変異p53細胞特異的治療法185) (後に必ずしもp53に依存しないことが判明)
は,p53が機能する正常細胞ではウイルスが複製できないが,p53に機能障害があるがん細胞では複製・増殖により細胞死に至る186,187).食道癌,肝癌に臨床第I相試験が,大腸癌,卵巣癌に臨床第II相試験が,頭頸部扁平上皮癌に臨床第III相試験行われている.感冒様症状のほかは重篤な有害事象は現時点では認められていない.抗がん剤との併用での応用が期待されている.
p53-MDM2結合阻害
p53の機能調節には,MDM2をはじめとするタンパク質結合によるものと,リン酸化,アセチル化などの翻訳後修飾によるものがある.このうちMDM2の結合はp53の機能調節のなかで最も重要なものと考えられる.MDM2のp53に対する機能は主に3つある.すなわち,1)
MDM2結合によりp53の塩基配列特異的転写活性化能を阻害する188,189),2)
p53の核外移送を促進する190),3) 自身のユビキチン・リガーゼ活性によりp53をユビキチン化してプロテアソームでの分解を促進する16).通常,細胞内のp53タンパク質量はこのMDMにより低レベルに抑制されている.従ってMDM2とp53の結合を阻害して細胞内のp53レベルを上げかつ転写抑制を解除することにより,がん細胞の増殖抑制とアポトーシス誘導を促進できるのでは,と1990年代後半に考えられるようになった.出芽酵母のtwo
hybridシステムなどの研究により,MDM2のN末端 (第19-102残基) は,p53のN末端 (第1-41残基) の転写活性化ドメインに結合することが明らかになっている188).また,X線結晶により,MDM2とp53の結合構造は解析されている191).このうち,MDM2
(17-125) とp53 (15-29) の結合はX線結晶構造解析では,p53がMDM2の溝に入り込み結合することが明らかにされている192).この構造を基に,MDM2とp53の結合を特異的に阻害する小分子化合物の開発が可能になった.これまでに,コンピューターを用いたin
silico screeningで1,4-benzodiazepine-2-one188) が,in
vitro実験系では,微生物抽出物のELISAによるスクリーニングから真菌代謝産物のchlorfusin193),などが報告されている.また,MDM2に結合するp53は立体構造上連続する15ペプチドであるため,ペプチドによる結合阻害も可能であると考えられた.合成ペプチドとアラニン置換によりp53の18-23残基がMDM2結合に必要な最小単位であることが判明し184),この部位に特異的に結合するペプチドが探索されている.ファージ・ディスプレイ法により野生型12残基ペプチド
(p53の16-27残基) よりも29倍結合が強い12残基ペプチド (F19, W23, L26の3残基のみ野生型と一致) の開発194),さらに,人工アミノ酸導入により,最大で1,740倍強力なペプチド阻害剤が開発されている195).これらのMDM2-p53結合阻害物質は,in
vitroでMDM2高発現細胞株 (p53野生型) の増殖を抑制し,下流遺伝子 (WAF1やMDM2)
の転写を活性化し,さらにcaspase 3の活性化を介したアポトーシス誘導が確認されている196).また,ペプチド阻害剤の場合,これらの効果が,正常細胞由来株やMDM2を高発現していないp53野生型株では少なかったことから,予想どおりMDM2-p53結合阻害によるアポトーシス誘導法は,MDM2高発現のがん細胞に特異性があると考えられる196).いずれも前臨床試験の段階である.最近,MDM2-p53結合阻害物質を合成化合物からスクリーニングした結果,IC50
が0.09 μMのシス・イミダゾール系化合物,Nutlin-3 (Roche) が発見された197).この化合物は,MDM2との共結晶構造解析により特異的結合が示されたほか,in
vitroでの活性やヌード・マウス移植腫瘍に対する抗腫瘍効果が示されている.今後,p53-MDM2結合阻害のよる新しい分子標的治療薬の開発に発展する可能性があり,注目されている.
変異p53の機能回復薬剤
ミスセンス変異によって失われたp53の機能を回復させることによりがん細胞の増殖を抑制する小分子化合物の開発が行われている.Rastinejadらのグループは,腫瘍に高頻度で見つかる一部の変異p53の抗原性をin
vitroで野生型に回復することを指標にして,10万種類以上の化合物スクリーニングからCP-31398 (Pfizer) を見出し,この化合物がin
vitroで変異p53発現細胞のp21WAF1遺伝子の転写活性化能を回復させるほか,in
vivoではヌード・マウス移植腫瘍に対して抗腫瘍効果を示している198).後に,CP-31398は高濃度でユビキチン化を介して野生型p53を安定化する作用が認められ199),この際,DNA傷害時とは異なり,p53のリン酸化は不要であることが示されている.一方,PRIMA-1は変異p53を誘導発現するヒト腫瘍細胞株を用いた化合物ライブラリーのスクリーニングによって見出され,アポトーシス誘導やin
vivoではヌード・マウス移植腫瘍に対する抗腫瘍効果が示されている200).さらに,このような活性を持つ薬剤としてellipticineとその誘導体が報告されている201).逆にPifithrinはp53機能を抑制する202)が,この化合物を投与されたマウスは致死量の放射線照射に耐性であり203),化学療法も含めて204) がん治療の際に正常細胞を保護するために役立つ可能性がある.
おわりに
細胞内のp53経路の機能障害は,DNA傷害後の細胞周期チェックポイント機構やアポトーシス誘導機構の破綻を生じ,発がんやがんの進展に関わると考えられる.p53経路には多くの遺伝子が活性化,翻訳後修飾,安定化,細胞内局在,転写下流遺伝子などに関与するが,p53経路の破綻は主にTP53遺伝子変異によるため,p53はがん抑制に関してp53経路のネットワークの中心的役割を果たすと考えられる.我々の1塩基置換モデルによると,ミスセンス変異によるp53の塩基配列特異的転写活性化能の抑制スペクトラムは腫瘍由来の変異スペクトラムとよく相関した.このことは塩基配列特異的転写活性化能がp53のがん抑制に重要な機能であり,この機能がTP53ミスセンス変異に対して感受性が高いことがTP53遺伝子変異の多くがミスセンス変異である理由であると推測される.しかしながら,TP53遺伝子変異でも変異の種類により報告頻度が異なり,機能的に多様性があると考えられる.また,細胞のがん抑制や治療感受性,個体の運命
(予後) はp53経路の破綻の原因 (TP53遺伝子変異とそれ以外の分子機序),TP53遺伝子多型の違い,p53ファミリーの発現によって異なる可能性がある.治療の対象となる腫瘍組織においてどのp53標的遺伝子がアポトーシス誘導に重要であるか,さらにどのTP53変異がその標的遺伝子の転写活性化やp73の機能を阻害するのか,また,どの抗がん剤ならTP53変異や特定遺伝子多型によって治療抵抗性とならないのか,今後明らかにすべき点も多い.TP53変異による細胞の変化のゲノム,トランスクリプトームおよびプロテオーム解析によりp53経路の破綻が細胞の運命に及ぼす影響を網羅的に解析し,p53経路に関連する新しい癌治療の分子標的の発見が期待される.この総説は,主にがん研究入門者または臨床腫瘍学を志す若手医師対象にp53とヒトがんの関連について解説した.
謝辞
変異p53ライブラリーの作成とそれを用いた研究は主に韓 双印,劉 文,加藤俊介,大塚和令,角道祐一,柴田浩行の各博士とともに行われました.ここに感謝の意を表します.
文 献
1) el-Deiry W, Kern S, Pietenpol J, Kinzler K, Vogelstein B. Definition of
a consensus binding site for p53. Nat Genet 1 : 45-49, 1992.
2) Vogelstein B, Kinzler K. p53 function and dysfunction. Cell 70 :
523-526, 1992.
3) Lane DP. Cancer. p53, guardian of the genome. Nature 358 :
15-16, 1992.
4) Ko L, Prives C. p53:puzzle and paradigm. Genes Dev 10 : 1054-1072,
1996.
5) Levine A. p53, the cellular gatekeeper for growth and division. Cell 88 :
323-331, 1997.
6) Velculescu VE, El-Deiry WS. Biological and clinical importance of the p53
tumor suppressor gene. Clin Chem 42 : 858-868, 1996.
7) Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 408 :
307-310, 2000.
8) Vousden KH, Lu X. Live or let die:the cell's response to p53. Nat Rev Cancer 2 :
594-604, 2002.
9) Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf:progress and puzzles. Curr
Opin Genet Dev 13 : 77-83, 2003.
10) Hussain SP, Harris CC. Molecular epidemiology of human cancer:contribution
of mutation spectra studies of tumor suppressor genes. Cancer Res 58 :
4023-4037, 1998.
11) Beroud C, Soussi T. The UMD-p53 database:new mutations and analysis tools. Hum
Mutat 21 : 176-181, 2003.
12) Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene
product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69 :
1237-1245, 1992.
13) Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification
of a gene encoding a p53-associated protein in human sarcomas. Nature 358 :
80-83, 1992.
14) Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein
encoded by human papillomavirus types 16 and 18 promotes the degradation of
p53. Cell 63 : 1129-1136, 1990.
15) Maki CG, Huibregtse JM, Howley PM. In vivo ubiquitination and proteasome-mediated
degradation of p53(1). Cancer Res 56 : 2649-2654, 1996.
16) Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3
for tumor suppressor p53. FEBS Lett 420 : 25-27, 1997.
17) Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 387 :
299-303, 1997.
18) Irwin MS, Kaelin WG, Jr. Role of the newer p53 family proteins in malignancy. Apoptosis 6 :
17-29, 2001.
19) Nakamura Y. Isolation of p53-target genes and their functional analysis. Cancer
Sci 95 : 7-11, 2004.
20) Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H,
Tamai K, Tokino T, Nakamura Y, Taya Y. p53AIP1, a potential mediator of p53-dependent
apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 102 :
849-862, 2000.
21) Samuels-Lev Y, O'Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong
S, Campargue I, Naumovski L, Crook T, Lu X. ASPP proteins specifically stimulate
the apoptotic function of p53. Mol Cell 8 : 781-794, 2001.
22) Moll UM, Zaika A. Nuclear and mitochondrial apoptotic pathways of p53. FEBS
Lett 493 : 65-69, 2001.
23) Soussi T, Beroud C. Assessing TP53 status in human tumours to evaluate
clinical outcome. Nat Rev Cancer 1 : 233-240, 2001.
24) El-Deiry WS. The role of p53 in chemosensitivity and radiosensitivity. Oncogene 22 :
7486-7495, 2003.
25) Morgan SE, Kastan MB. p53 and ATM:cell cycle, cell death, and cancer. Adv
Cancer Res 71 : 1-25, 1997.
26) Westphal CH. Cell-cycle signaling:Atm displays its many talents. Curr Biol 7 :
R789-792, 1997.
27) Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB. DNA
damage induces phosphorylation of the amino terminus of p53. Genes Dev 11 :
3471-3481, 1997.
28) Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY,
Taya Y, Prives C, Abraham RT. A role for ATR in the DNA damage-induced phosphorylation
of p53. Genes Dev 13 : 152-157, 1999.
29) Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint
kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible
sites. Genes Dev 14 : 289-300, 2000.
30) Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge
SJ, Mak TW. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287 :
1824-1827, 2000.
31) Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as
a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev 14 :
278-288, 2000.
32) O'Hagan HM, Ljungman M. Nuclear accumulation of p53 following inhibition
of transcription is not due to diminished levels of MDM2. Oncogene 23 :
5505-5512, 2004.
33) Shmueli A, Oren M. Regulation of p53 by Mdm2:fate is in the numbers. Mol
Cell 13 : 4-5, 2004.
34) Lane D, Lain S. Therapeutic exploitation of the p53 pathway. Trends Mol
Med 8 : S38-42, 2002.
35) el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin
D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor
suppression. Cell 75 : 817-825, 1993.
36) Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, Love DR,
Mole SE, Moore JK, Papi L, et al. Germ-line mutations of the RET proto-oncogene
in multiple endocrine neoplasia type 2A. Nature 363 : 458-460,
1993.
37) Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, Scherer SW, Zhuang
Z, Lubensky I, Dean M, Allikmets R, Chidambaram A, Bergerheim UR, Feltis JT,
Casadevall C, Zamarron A, Bernues M, Richard S, Lips CJ, Walther MM, Tsui LC,
Geil L, Orcutt ML, Stackhouse T, Zbar B, et al. Germline and somatic mutations
in the tyrosine kinase domain of the MET proto-oncogene in papillary renal
carcinomas. Nat Genet 16 : 68-73, 1997.
38) Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, Kanakura
Y, Tanaka T, Takabayashi A, Matsuda H, Kitamura Y. Familial gastrointestinal
stromal tumours with germline mutation of the KIT gene. Nat Genet 19 :
323-324, 1998.
39) Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja
TP. A human DNA segment with properties of the gene that predisposes to retinoblastoma
and osteosarcoma. Nature 323 : 643-646, 1986.
40) Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M,
Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary
nonpolyposis colon cancer. Cell 75 : 1027-1038, 1993.
41) Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomaki
P, Sistonen P, Aaltonen LA, Nystrom-Lahti M, et al. Mutations of a mutS homolog
in hereditary nonpolyposis colorectal cancer. Cell 75 : 1215-1225,
1993.
42) Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, Kane
M, Earabino C, Lipford J, Lindblom A, et al. Mutation in the DNA mismatch repair
gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 368 :
258-261, 1994.
43) Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, Haseltine
WA, Fleischmann RD, Fraser CM, Adams MD, et al. Mutation of a mutL homolog
in hereditary colon cancer. Science 263 : 1625-1629, 1994.
44) Malkin D, Li FP, Strong LC, Fraumeni JF, Jr, Nelson CE, Kim DH, Kassel
J, Gryka MA, Bischoff FZ, Tainsky MA, et al. Germ line p53 mutations in a familial
syndrome of breast cancer, sarcomas, and other neoplasms. Science 250 :
1233-1238, 1990.
45) Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich
M, Verselis SJ, Isselbacher KJ, Fraumeni JF, Birch JM, Li FP, Garber JE, Haber
DA. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 286 :
2528-2531, 1999.
46) Korsmeyer SJ. Bcl-2:an antidote to programmed cell death. Cancer Surv 15 :
105-118, 1992.
47) Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS, Santee SM, Fujiwara T, Roth
JA, Deisseroth AB, Zhang WW, Kruzel E, et al. Wild-type human p53 and a temperature-sensitive
mutant induce Fas/APO-1 expression. Mol Cell Biol 15 : 3032-3040,
1995.
48) Wu GS, Burns TF, McDonald ER, 3rd, Jiang W, Meng R, Krantz ID, Kao G, Gan
DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G, el-Deiry
WS. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat
Genet 17 : 141-143, 1997.
49) Maecker HL, Koumenis C, Giaccia AJ. p53 promotes selection for Fas-mediated
apoptotic resistance. Cancer Res 60 : 4638-4644, 2000.
50) Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional
activator of the human bax gene. Cell 80 : 293-299, 1995.
51) Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol
Cell 7 : 683-694, 2001.
52) Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi
T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator
of p53-induced apoptosis. Science 288 : 1053-1058, 2000.
53) Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS. BID regulation
by p53 contributes to chemosensitivity. Nat Cell Biol 4 : 842-849,
2002.
54) Kannan K, Kaminski N, Rechavi G, Jakob-Hirsch J, Amariglio N, Givol D. DNA
microarray analysis of genes involved in p53 mediated apoptosis:activation
of Apaf-1. Oncogene 20 : 3449-3455, 2001.
55) MacLachlan TK, El-Deiry WS. Apoptotic threshold is lowered by p53 transactivation
of caspase-6. Proc Natl Acad Sci USA 99 : 9492-9497. Epub 2002
Jun 9427, 2002.
56) Attardi LD, Reczek EE, Cosmas C, Demicco EG, McCurrach ME, Lowe SW, Jacks
T. PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3
family. Genes Dev 14 : 704-718, 2000.
57) Lin Y, Ma W, Benchimol S. Pidd, a new death-domain-containing protein,
is induced by p53 and promotes apoptosis. Nat Genet 26 : 122-127,
2000.
58) Okamura S, Arakawa H, Tanaka T, Nakanishi H, Ng CC, Taya Y, Monden M, Nakamura
Y. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol Cell 8 :
85-94, 2001.
59) Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak
TW. Regulation of PTEN transcription by p53. Mol Cell 8 : 317-325,
2001.
60) Mack DH, Vartikar J, Pipas JM, Laimins LA. Specific repression of TATA-mediated
but not initiator-mediated transcription by wild-type p53. Nature 363 :
281-283, 1993.
61) Zhang CC, Yang JM, Bash-Babula J, White E, Murphy M, Levine AJ, Hait WN. DNA
damage increases sensitivity to vinca alkaloids and decreases sensitivity to
taxanes through p53-dependent repression of microtubule-associated protein
4. Cancer Res 59 : 3663-3670, 1999.
62) Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression
of the anti-apoptotic survivin gene by wild type p53. J Biol Chem 277 :
3247-3257. Epub 2001 Nov 3219, 2002.
63) Koumenis C, Alarcon R, Hammond E, Sutphin P, Hoffman W, Murphy M, Derr
J, Taya Y, Lowe SW, Kastan M, Giaccia A. Regulation of p53 by hypoxia:dissociation
of transcriptional repression and apoptosis from p53-dependent transactivation. Mol
Cell Biol 21 : 1297-1310, 2001.
64) Caelles C, Helmberg A, Karin M. p53-dependent apoptosis in the absence
of transcriptional activation of p53-target genes. Nature 370 :
220-223, 1994.
65) Wagner AJ, Kokontis JM, Hay N. Myc-mediated apoptosis requires wild-type
p53 in a manner independent of cell cycle arrest and the ability of p53 to
induce p21waf1/cip1. Genes Dev 8 : 2817-2830, 1994.
66) Haupt Y, Rowan S, Shaulian E, Vousden KH, Oren M. Induction of apoptosis
in HeLa cells by trans-activation-deficient p53. Genes Dev 9 :
2170-2183, 1995.
67) Chen X, Ko LJ, Jayaraman L, Prives C. p53 levels, functional domains, and
DNA damage determine the extent of the apoptotic response of tumor cells. Genes
Dev 10 : 2438-2451, 1996.
68) Kokontis JM, Wagner AJ, O'Leary M, Liao S, Hay N. A transcriptional activation
function of p53 is dispensable for and inhibitory of its apoptotic function. Oncogene 20 :
659-668, 2001.
69) Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M,
Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization
and apoptosis. Science 303 : 1010-1014, 2004.
70) Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll
UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11 :
577-590, 2003.
71) Dumont P, Leu JI, Della Pietra AC, 3rd, George DL, Murphy M. The codon
72 polymorphic variants of p53 have markedly different apoptotic potential. Nat
Genet 33 : 357-365, 2003. 72) Resnick MA, Inga A. Functional mutants
of the sequence-specific transcription factor p53 and implications for master
genes of diversity. Proc Natl Acad Sci USA 100 : 9934-9939. Epub
2003 Aug 9938, 2003.
73) Zhao R, Gish K, Murphy M, Yin Y, Notterman D, Hoffman WH, Tom E, Mack DH,
Levine AJ. Analysis of p53-regulated gene expression patterns using oligonucleotide
arrays. Genes Dev 14 : 981-993, 2000.
74) Ashcroft M, Kubbutat M, Vousden K. Regulation of p53 function and stability
by phosphorylation. Mol Cell Biol 19 : 1751-1758, 1999.
75) Barlev N, Liu L, Chehab N, Mansfield K, Harris K, Halazonetis T, Berger
S. Acetylation of p53 activates transcription through recruitment of coactivators/histone
acetyltransferases. Mol Cell 8 : 1243-1254, 2001.
76) Rodriguez MS, Desterro JM, Lain S, Midgley CA, Lane DP, Hay RT. SUMO-1
modification activates the transcriptional response of p53. Embo J 18 :
6455-6461, 1999.
77) Gostissa M, Hengstermann A, Fogal V, Sandy P, Schwarz SE, Scheffner M,
Del Sal G. Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. Embo
J 18 : 6462-6471, 1999.
78) Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S, Ronai
Z, Blandino G, Schneider C, Del SG. The prolyl isomerase Pin1 reveals a mechanism
to control p53 functions after genotoxic insults. Nature 419 :
853-857, 2002.
79) Zheng H, You H, Zhou X, Murray S, Uchida T, Wulf G, Gu L, Tang X, Lu K,
Xiao Z. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 419 :
849-853, 2002.
80) Nigro J, Baker S, Preisinger A, Jessup J, Hostetter R, Cleary K, Bigner
S, Davidson N, Baylin S, Devilee P, et al. Mutations in the p53 gene occur
in diverse human tumour types. Nature 342 : 705-708, 1989.
81) Soussi T, Dehouche K, Beroud C. p53 website and analysis of p53 gene mutations
in human cancer:forging a link between epidemiology and carcinogenesis. Hum
Mutat 15 : 105-113, 2000.
82) Olivier M, Eeles R, Hollstein M, Khan M, Harris C, Hainaut P. The IARC
TP53 database:new online mutation analysis and recommendations to users. Hum
Mutat 19 : 607-614, 2002.
83) Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C. Understanding
the function-structure and function-mutation relationships of p53 tumor suppressor
protein by high-resolution missense mutation analysis. Proc Natl Acad Sci USA 100 :
8424-8429, 2003.
84) De Vries EM, Ricke DO, De Vries TN, Hartmann A, Blaszyk H, Liao D, Soussi
T, Kovach JS, Sommer SS. Database of mutations in the p53 and APC tumor suppressor
genes designed to facilitate molecular epidemiological analyses. Hum Mutat 7 :
202-213, 1996.
85) Harris CC. 1995 Deichmann Lecture―p53 tumor suppressor gene:at the crossroads
of molecular carcinogenesis, molecular epidemiology and cancer risk assessment. Toxicol
Lett 82-83 : 1-7, 1995.
86) Hussain SP, Harris CC. Molecular epidemiology and carcinogenesis:endogenous
and exogenous carcinogens. Mutat Res 462 : 311-322, 2000.
87) Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor
suppressor-DNA complex:understanding tumorigenic mutations. Science 265 :
346-355, 1994.
88) Clore GM, Omichinski JG, Sakaguchi K, Zambrano N, Sakamoto H, Appella E,
Gronenborn AM. High-resolution structure of the oligomerization domain of p53
by multidimensional NMR. Science 265 : 386-391, 1994.
89) Jeffrey PD, Gorina S, Pavletich NP. Crystal structure of the tetramerization
domain of the p53 tumor suppressor at 1.7 angstroms. Science 267 :
1498-1502, 1995.
90) Ishioka C, Frebourg T, Yan YX, Vidal M, Friend SH, Schmidt S, Iggo R. Screening
patients for heterozygous p53 mutations using a functional assay in yeast. Nature
Genetics 5 : 124-129, 1993.
91) Bargonetti J, Manfredi JJ, Chen X, Marshak DR, Prives C. A proteolytic
fragment from the central region of p53 has marked sequence-specific DNA-binding
activity when generated from wild -type but not from oncogenic mutant p53 protein. Genes
Dev 7 : 2565-2574, 1993.
92) Pavletich NP, Chambers KA, Pabo CO. The DNA-binding domain of p53 contains
the four conserved regions and the major mutation hot spots. Genes Dev 7 :
2556-2564, 1993.
93) Varley JM, McGown G, Thorncroft M, Cochrane S, Morrison P, Woll P, Kelsey
AM, Mitchell EL, Boyle J, Birch JM, Evans DG. A previously undescribed mutation
within the tetramerisation domain of TP53 in a family with Li-Fraumeni syndrome. Oncogene 12 :
2437-2442, 1996.
94) Ribeiro RC, Sandrini F, Figueiredo B, Zambetti GP, Michalkiewicz E, Lafferty
AR, DeLacerda L, Rabin M, Cadwell C, Sampaio G, Cat I, Stratakis CA, Sandrini
R. An inherited p53 mutation that contributes in a tissue-specific manner to
pediatric adrenal cortical carcinoma. Proc Natl Acad Sci USA 98 : 9330-9335,
2001.
95) Soussi T, Caron dFC, May P. Structural aspects of the p53 protein in relation
to gene evolution. Oncogene 5 : 945-952, 1990.
96) Liu G, McDonnell TJ, Montes de Oca Luna R, Kapoor M, Mims B, El-Naggar
AK, Lozano G. High metastatic potential in mice inheriting a targeted p53 missense
mutation. Proc Natl Acad Sci USA 97 : 4174-4179, 2000.
97) Birch JM, Blair V, Kelsey AM, Evans DG, Harris M, Tricker KJ, Varley JM. Cancer
phenotype correlates with constitutional TP53 genotype in families with the
Li-Fraumeni syndrome. Oncogene 17 : 1061-1068, 1998.
98) Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P, Eeles
RA. Li-Fraumeni and related syndromes:correlation between tumor type, family
structure, and TP53 genotype. Cancer Res 63 : 6643-6650, 2003.
99) Shiraishi K, Kato S, Han SY, Liu W, Otsuka K, Sakayori M, Ishida T, Takeda
M, Kanamaru R, Ohuchi N, Ishioka C. Isolation of temperature-sensitive p53
mutations from a comprehensive missense mutation library. J Biol Chem 279 :
348-355. Epub 2003 Oct 2013, 2004.
100) Halevy O, Michalovitz D, Oren M. Different tumor-derived p53 mutants exhibit
distinct biological activities. Science 250 : 113-116, 1990.
101) Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, Finlay C, Levine
AJ. Gain of function mutations in p53. Nat Genet 4 : 42-46, 1993.
102) Harvey M, Vogel H, Morris D, Bradley A, Bernstein A, Donehower LA. A mutant
p53 transgene accelerates tumour development in heterozygous but not nullizygous
p53-deficient mice. Nat Genet 9 : 305-311, 1995.
103) Brachmann RK, Vidal M, Boeke JD. Dominant-negative p53 mutations selected
in yeast hit cancer hot spots. Proc Natl Acad Sci USA 93 : 4091-4095,
1996.
104) Walker KK, Levine AJ. Identification of a novel p53 functional domain
that is necessary for efficient growth suppression. Proc Natl Acad Sci USA 93 :
15335-15340, 1996.
105) Blandino G, Levine AJ, Oren M. Mutant p53 gain of function:differential
effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 18 :
477-485, 1999.
106) Soussi T, Kato S, P L, Ishioka C. Reassessment of the TP53 mutation datab
ase in human disease by data mining with a library of TP53 missense mutations. Hum
Mutat (in press), 2004.
107) Tornaletti S, Pfeifer GP. Complete and tissue-independent methylation
of CpG sites in the p53 gene:implications for mutations in human cancers. Oncogene 10 :
1493-1499, 1995.
108) Denissenko MF, Pao A, Tang M, Pfeifer GP. Preferential formation of benzo
[a] pyrene adducts at lung cancer mutational hotspots in P53. Science 274 :
430-432, 1996.
109) Rodin SN, Rodin AS. Human lung cancer and p53:the interplay between mutagenesis
and selection. Proc Natl Acad Sci USA 97 : 12244-12249, 2000.
110) Mitsudomi T, Hamajima N, Ogawa M, Takahashi T. Prognostic significance
of p53 alterations in patients with non-small cell lung cancer:a meta-analysis. Clin
Cancer Res 6 : 4055-4063, 2000.
111) Huncharek M, Kupelnick B, Geschwind JF, Caubet JF. Prognostic significance
of p53 mutations in non-small cell lung cancer:a meta-analysis of 829 cases
from eight published studies. Cancer Lett 153 : 219-226, 2000.
112) Steels E, Paesmans M, Berghmans T, Branle F, Lemaitre F, Mascaux C, Meert
AP, Vallot F, Lafitte JJ, Sculier JP. Role of p53 as a prognostic factor for
survival in lung cancer:a systematic review of the literature with a meta-analysis. Eur
Respir J 18 : 705-719, 2001.
113) Pharoah PD, Day NE, Caldas C. Somatic mutations in the p53 gene and prognosis
in breast cancer:a meta-analysis. Br J Cancer 80 : 1968-1973, 1999.
114) Iacopetta B. TP53 mutation in colorectal cancer. Hum Mutat 21 :
271-276, 2003.
115) Schmitz-Drager BJ, Goebell PJ, Ebert T, Fradet Y. p53 immunohistochemistry
as a prognostic marker in bladder cancer. Playground for urology scientists? Eur Urol 38 : 691-699;discussion 700, 2000.
116) Moreno Sierra J, Fernandez Perez C, Mazuecos Lopez MP, Lopez Garcia J,
Barreales Tolosa L, Fernandez Montarroso L, Lopez Corral JC, Blanco Jimenez
E, Silmi Moyano A. [Systematic review about the usefulness and prognostic value
of the p53 oncoprotein and proliferation marker Ki-67 for upper urinary tract
transitional cell carcinoma]. Arch Esp Urol 57 : 327-335, 2004.
117) Kmet LM, Cook LS, Magliocco AM. A review of p53 expression and mutation
in human benign, low malignant potential, and invasive epithelial ovarian tumors. Cancer 97 :
389-404, 2003.
118) Nieder C, Petersen S, Petersen C, Thames HD. The challenge of p53 as prognostic
and predictive factor in gliomas. Cancer Treat Rev 26 : 67-73, 2000.
119) Aas T, Borresen AL, Geisler S, Smith-Sorensen B, Johnsen H, Varhaug JE,
Akslen LA, Lonning PE. Specific P53 mutations are associated with de novo resistance
to doxorubicin in breast cancer patients. Nat Med 2 : 811-814, 1996.
120) Borresen AL, Andersen TI, Eyfjord JE, Cornelis RS, Thorlacius S, Borg
A, Johansson U, Theillet C, Scherneck S, Hartman S, et al. TP53 mutations and
breast cancer prognosis:particularly poor survival rates for cases with mutations
in the zinc-binding domains. Genes Chromosomes Cancer 14 : 71-75, 1995.
121) Berns EM, van Staveren IL, Look MP, Smid M, Klijn JG, Foekens JA. Mutations
in residues of TP53 that directly contact DNA predict poor outcome in human
primary breast cancer. Br J Cancer 77 : 1130-1136, 1998.
122) Gentile M, Bergman Jungestrom M, Olsen KE, Soderkvist P, Wingren S. p53
and survival in early onset breast cancer:analysis of gene mutations, loss
of heterozygosity and protein accumulation. Eur J Cancer 35 : 1202-1207,
1999.
123) Geisler S, Lonning PE, Aas T, Johnsen H, Fluge O, Haugen DF, Lillehaug
JR, Akslen LA, Borresen-Dale AL. Influence of TP53 gene alterations and c-erbB-2
expression on the response to treatment with doxorubicin in locally advanced
breast cancer. Cancer Res 61 : 2505-2512, 2001.
124) Powell B, Soong R, Iacopetta B, Seshadri R, Smith DR. Prognostic significance
of mutations to different structural and functional regions of the p53 gene
in breast cancer. Clin Cancer Res 6 : 443-451, 2000.
125) Skaug V, Ryberg D, Kure EH, Arab MO, Stangeland L, Myking AO, Haugen A. p53
mutations in defined structural and functional domains are related to poor
clinical outcome in non-small cell lung cancer patients. Clin Cancer Res 6 :
1031-1037, 2000.
126) Kihara C, Seki T, Furukawa Y, Yamana H, Kimura Y, van Schaardenburgh P,
Hirata K, Nakamura Y. Mutations in zinc-binding domains of p53 as a prognostic
marker of esophageal-cancer patients. Jpn J Cancer Res 91 : 190-198, 2000.
127) Borresen-Dale AL, Lothe RA, Meling GI, Hainaut P, Rognum TO, Skovlund
E. TP53 and long-term prognosis in colorectal cancer:mutations in the L3 zinc-binding
domain predict poor survival. Clin Cancer Res 4 : 203-210, 1998.
128) Erber R, Conradt C, Homann N, Enders C, Finckh M, Dietz A, Weidauer H,
Bosch FX. TP53 DNA contact mutations are selectively associated with allelic
loss and have a strong clinical impact in head and neck cancer. Oncogene 16 :
1671-1679, 1998.
129) Tomizawa Y, Kohno T, Fujita T, Kiyama M, Saito R, Noguchi M, Matsuno Y,
Hirohashi S, Yamaguchi N, Nakajima T, Yokota J. Correlation between the status
of the p53 gene and survival in patients with stage I non-small cell lung carcinoma. Oncogene
18 : 1007-1014, 1999.
130) Goh HS, Yao J, Smith DR. p53 point mutation and survival in colorectal
cancer patients. Cancer Res 55 : 5217-5221, 1995.
131) Samowitz WS, Curtin K, Ma KN, Edwards S, Schaffer D, Leppert MF, Slattery
ML. Prognostic significance of p53 mutations in colon cancer at the population
level. Int J Cancer 99 : 597-602, 2002.
132) Storey A, Thomas M, Kalita A, Harwood C, Gardiol D, Mantovani F, Breuer
J, Leigh IM, Matlashewski G, Banks L. Role of a p53 polymorphism in the development
of human papillomavirus-associated cancer. Nature 393 : 229-234, 1998.
133) Koushik A, Platt RW, Franco EL. p53 codon 72 polymorphism and cervical
neoplasia:a meta-analysis review. Cancer Epidemiol Biomarkers Prev 13 :
11-22, 2004.
134) Huang XE, Hamajima N, Katsuda N, Matsuo K, Hirose K, Mizutani M, Iwata
H, Miura S, Xiang J, Tokudome S, Tajima K. Association of p53 codon Arg72Pro
and p73 G4C14-to-A4T14 at exon 2 genetic polymorphisms with the risk of Japanese
breast cancer. Breast Cancer 10 : 307-311, 2003.
135) Gemignani F, Moreno V, Landi S, Moullan N, Chabrier A, Gutierrez-Enriquez
S, Hall J, Guino E, Peinado MA, Capella G, Canzian F. A TP53 polymorphism is
associated with increased risk of colorectal cancer and with reduced levels
of TP53 mRNA. Oncogene 23 : 1954-1956, 2004.
136) Wu HC, Chang CH, Chen HY, Tsai FJ, Tsai JJ, Chen WC. p53 gene codon 72
polymorphism but not tumor necrosis factor-alpha gene is associated with prostate
cancer. Urol Int 73 : 41-46, 2004.
137) Granja F, Morari J, Morari EC, Correa LA, Assumpcao LV, Ward LS. Proline
homozygosity in codon 72 of p53 is a factor of susceptibility for thyroid cancer. Cancer
Lett 210 : 151-157, 2004.
138) Kuroda Y, Tsukino H, Nakao H, Imai H, Katoh T. p53 Codon 72 polymorphism
and urothelial cancer risk. Cancer Lett 189 : 77-83, 2003.
139) B ergamaschi G, Merante S, Orlandi E, Galli A, Bernasconi P, Cazzola M. TP53
codon 72 polymorphism in patients with chronic myeloid leukemia. Haematologica
89 : 868-869, 2004.
140) Matakidou A, Eisen T, Houlston RS. TP53 polymorphisms and lung cancer
risk:a systematic review and meta-analysis. Mutagenesis 18 : 377-385, 2003.
141) Marin MC, Jost CA, Brooks LA, Irwin MS, O'Nions J, Tidy JA, James N, McGregor
JM, Harwood CA, Yulug IG, Vousden KH, Allday MJ, Gusterson B, Ikawa S, Hinds
PW, Crook T, Kaelin WG, Jr. A common polymorphism acts as an intragenic modifier
of mutant p53 behaviour. Nat Genet 25 : 47-54, 2000.
142) Langerod A, Bukholm IR, Bregard A, Lonning PE, Andersen TI, Rognum TO,
Meling GI, Lothe RA, Borresen-Dale AL. The TP53 codon 72 polymorphism may affect
the function of TP53 mutations in breast carcinomas but not in colorectal carcinomas. Cancer
Epidemiol Biomarkers Prev 11 : 1684-1688, 2002.
143) Papadakis ED, Soulitzis N, Spandidos DA. Association of p53 codon 72 polymorphism
with advanced lung cancer:the Arg allele is preferentially retained in tumours
arising in Arg/Pro germline heterozygotes. Br J Cancer 87 : 1013-1018,
2002.
144) Furihata M, Takeuchi T, Matsumoto M, Kurabayashi A, Ohtsuki Y, Terao N,
Kuwahara M, Shuin T. p53 mutation arising in Arg72 allele in the tumorigenesis
and development of carcinoma of the urinary tract. Clin Cancer Res 8 :
1192-1195, 2002.
145) Tada M, Furuuchi K, Kaneda M, Matsumoto J, Takahashi M, Hirai A, Mitsumoto
Y, Iggo RD, Moriuchi T. Inactivate the remaining p53 allele or the alternate
p73? Preferential selection of the Arg72 polymorphism in cancers with recessive
p53 mutants but not transdominant mutants. Carcinogenesis 22 : 515-517,
2001.
146) Wang YC, Lee HS, Chen SK, Chang YY, Chen CY. Prognostic significance of
p53 codon 72 polymorphism in lung carcinomas. Eur J Cancer 35 : 226-230,
1999.
147) Bonafe M, Ceccarelli C, Farabegoli F, Santini D, Taffurelli M, Barbi C,
Marzi E, Trapassi C, Storci G, Olivieri F, Franceschi C. Retention of the p53
codon 72 arginine allele is associated with a reduction of disease-free and
overall survival in arginine/proline heterozygous breast cancer patients. Clin
Cancer Res 9 : 4860-4864, 2003.
148) O'Connor PM, Jackman J, Bae I, Myers TG, Fan S, Mutoh M, Scudiero DA,
Monks A, Sausville EA, Weinstein JN, Friend S, Fornace AJ, Jr, Kohn KW. Characterization
of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute
anticancer drug screen and correlations with the growth-inhibitory potency
of 123 anticancer agents. Cancer Res 57 : 4285-4300, 1997.
149) Weinstein JN, Myers TG, O'Connor PM, Friend SH, Fornace AJ, Jr, Kohn KW,
Fojo T, Bates SE, Rubinstein LV, Anderson NL, Buolamwini JK, van Osdol WW,
Monks AP, Scudiero DA, Sausville EA, Zaharevitz DW, Bunow B, Viswanadhan VN,
Johnson GS, Wittes RE, Paull KD. An information-intensive approach to the molecular
pharmacology of cancer. Science 275 : 343-349, 1997.
150) Geisler S, Borresen-Dale AL, Johnsen H, Aas T, Geisler J, Akslen LA, Anker
G, Lonning PE. TP53 gene mutations predict the response to neoadjuvant treatment
with 5-fluorouracil and mitomycin in locally advanced breast cancer. Clin Cancer
Res 9 : 5582-5588, 2003.
151) Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, Minty A,
Chalon P, Lelias JM, Dumont X, Ferrara P, McKeon F, Caput D. Monoallelically
expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma
and other human cancers. Cell 90 : 809-819, 1997.
152) Osada M, Ohba M, Kawahara C, Ishioka C, Kanamaru R, Katoh I, Ikawa Y,
Nimura Y, Nakagawara A, Obinata M, Ikawa S. Cloning and functional analysis
of human p51, which structurally and functionally resembles p53. Nat Med 4 :
839-843, 1998.
153) Kato S, Shimada A, Osada M, Ikawa S, Obinata M, Nakagawara A, Kanamaru
R, Ishioka C. Effects of p51/p63 missense mutations on transcriptional activities
of p53 downstream gene promoters. Cancer Res 59 : 5908-5911, 1999.
154) Han S, Semba S, Abe T, Makino N, Furukawa T, Fukushige S, Takahashi H,
Sakurada A, Sato M, Shiiba K, Matsuno S, Nimura Y, Nakagawara A, Horii A. Infrequent
somatic mutations of the p73 gene in various human cancers. Eur J Surg Oncol
25 : 194-198, 1999.
155) Hagiwara K, McMenamin MG, Miura K, Harris CC. Mutational analysis of the
p63/p73L/p51/p40/CUSP/KET gene in human cancer cell lines using intronic primers. Cancer
Res 59 : 4165-4169, 1999.
156) Kaelin WG, Jr. The emerging p53 gene family. J Natl Cancer Inst 91 :
594-598, 1999.
157) Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, Liu W, Flores ER,
Tsai KY, Jacks T, Vousden KH, Kaelin WG, Jr. Role for the p53 homologue p73
in E2F-1-induced apoptosis. Nature 407 : 645-648, 2000.
158) Melino G, Lu X, Gasco M, Crook T, Knight RA. Functional regulation of
p73 and p63:development and cancer. Trends Biochem Sci 28 : 663-670, 2003.
159) Shimada A, Kato S, Enjo K, Osada M, Ikawa Y, Kohno K, Obinata M, Kanamaru
R, Ikawa S, Ishioka C. The transcriptional activities of p53 and its homologue
p51/p63:similarities and differences. Cancer Res 59 : 2781-2786, 1999.
160) Jost CA, Marin MC, Kaelin WG, Jr. p73 is a simian [correction of human]
p53-related protein that can induce apoptosis. Nature 389 : 191-194,
1997.
161) Ishimoto O, Kawahara C, Enjo K, Obinata M, Nukiwa T, Ikawa S. Possible
oncogenic potential of DeltaNp73:a newly identified isoform of human p73. Cancer
Res 62 : 636-641, 2002.
162) Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic
role for the p53 family member, p73, during developmental neuron death. Science 289 :
304-306, 2000.
163) Stiewe T, Zimmermann S, Frilling A, Esche H, Putzer BM. Transactivation-deficient
DeltaTA-p73 acts as an oncogene. Cancer Res 62 : 3598-3602, 2002.
164) Zaika AI, Slade N, Erster SH, Sansome C, Joseph TW, Pearl M, Chalas E,
Moll UM. DeltaNp73, a dominant-negative inhibitor of wild-type p53 and TAp73,
is up-regulated in human tumors. J Exp Med 196 : 765-780, 2002.
165) Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T. p63
and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 416 :
560-564, 2002.
166) Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG, Jr, Levrero M, Wang
JY. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced
DNA damage. Nature 399 : 806-809, 1999.
167) Costanzo A, Merlo P, Pediconi N, Fulco M, Sartorelli V, Cole PA, Fontemaggi
G, Fanciulli M, Schiltz L, Blandino G, Balsano C, Levrero M. DNA damage-dependent
acetylation of p73 dictates the selective activation of apoptotic target genes. Mol
Cell 9 : 175-186, 2002.
168) Di Como CJ, Gaiddon C, Prives C. p73 function is inhibited by tumor-derived
p53 mutants in mammalian cells. Mol Cell Biol 19 : 1438-1449, 1999.
169) Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG, Jr. Chemosensitivity
linked to p73 function. Cancer Cell 3 : 403-410, 2003.
170) Sullivan A, Syed N, Gasco M, Bergamaschi D, Trigiante G, Attard M, Hiller
L, Farrell PJ, Smith P, Lu X, Crook T. Polymorphism in wild-type p53 modulates
response to chemotherapy in vitro and in vivo. Oncogene 23 : 3328-3337,
2004.
171) Bergamaschi D, Gasco M, Hiller L, Sullivan A, Syed N, Trigiante G, Yulug
I, Merlano M, Numico G, Comino A, Attard M, Reelfs O, Gusterson B, Bell AK,
Heath V, Tavassoli M, Farrell PJ, Smith P, Lu X, Crook T. p53 polymorphism
influences response in cancer chemotherapy via modulation of p73-dependent
apoptosis. Cancer Cell 3 : 387-402, 2003.
172) Burns TF, Bernhard EJ, El-Deiry WS. Tissue specific expression of p53
target genes suggests a key role for KILLER/DR5 in p53-dependent apoptosis
in vivo. Oncogene 20 : 4601-4612, 2001.
173) Fei P, Bernhard EJ, El-Deiry WS. Tissue-specific induction of p53 targets
in vivo. Cancer Res 62 : 7316-7327, 2002.
174) Blagosklonny MV, El-Deiry WS. Acute overexpression of wt p53 facilitates
anticancer drug-induced death of cancer and normal cells. Int J Cancer 75 :
933-940, 1998.
175) Lanni JS, Lowe SW, Licitra EJ, Liu JO, Jacks T. p53-independent apoptosis
induced by paclitaxel through an indirect mechanism. Proc Natl Acad Sci USA 94 :
9679-9683, 1997.
176) Lang FF, Bruner JM, Fuller GN, Aldape K, Prados MD, Chang S, Berger MS,
McDermott MW, Kunwar SM, Junck LR, Chandler W, Zwiebel JA, Kaplan RS, Yung
WK. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma:biological
and clinical results. J Clin Oncol 21 : 2508-2518, 2003.
177) Pagliaro LC, Keyhani A, Williams D, Woods D, Liu B, Perrotte P, Slaton
JW, Merritt JA, Grossman HB, Dinney CP. Repeated intravesical instillations
of an adenoviral vector in patients with locally advanced bladder cancer:a
phase I study of p53 gene therapy. J Clin Oncol 21 : 2247-2253,
2003.
178) Schuler M, Rochlitz C, Horowitz JA, Schlegel J, Perruchoud AP, Kommoss
F, Bolliger CT, Kauczor HU, Dalquen P, Fritz MA, Swanson S, Herrmann R, Huber
C. A phase I study of adenovirus-mediated wild-type p53 gene transfer in patients
with advanced non-small cell lung cancer. Hum Gene Ther 9 : 2075-2082,
1998.
179) Swisher SG, Roth JA, Nemunaitis J, Lawrence DD, Kemp BL, Carrasco CH,
Connors DG, El-Naggar AK, Fossella F, Glisson BS, Hong WK, Khuri FR, Kurie
JM, Lee JJ, Lee JS, Mack M, Merritt JA, Nguyen DM, Nesbitt JC, Perez-Soler
R, Pisters KM, Putnam JB, Jr, Richli WR, Savin M, Waugh MK, et al. Adenovirus-mediated
p53 gene transfer in advanced non-small-cell lung cancer. J Natl Cancer Inst 91 :
763-771, 1999.
180) Swisher SG, Roth JA, Komaki R, Gu J, Lee JJ, Hicks M, Ro JY, Hong WK,
Merritt JA, Ahrar K, Atkinson NE, Correa AM, Dolormente M, Dreiling L, El-Naggar
AK, Fossella F, Francisco R, Glisson B, Grammer S, Herbst R, Huaringa A, Kemp
B, Khuri FR, Kurie JM, Liao Z, McDonnell TJ, Morice R, Morello F, Munden R,
Papadimitrakopoulou V, Pisters KM, Putnam JB, Jr, Sarabia AJ, Shelton T, Stevens
C, Shin DM, Smythe WR, Vaporciyan AA, Walsh GL, Yin M. Induction of p53-regulated
genes and tumor regression in lung cancer patients after intratumoral delivery
of adenoviral p53 (INGN 201) and radiation therapy. Clin Cancer Res 9 :
93-101, 2003.
181) Clayman GL, Frank DK, Bruso PA, Goepfert H. Adenovirus-mediated wild-type
p53 gene transfer as a surgical adjuvant in advanced head and neck cancers. Clin
Cancer Res 5 : 1715-1722, 1999.
182) Schuler M, Herrmann R, De Greve JL, Stewart AK, Gatzemeier U, Stewart
DJ, Laufman L, Gralla R, Kuball J, Buhl R, Heussel CP, Kommoss F, Perruchoud
AP, Shepherd FA, Fritz MA, Horowitz JA, Huber C, Rochlitz C. Adenovirus-mediated
wild-type p53 gene transfer in patients receiving chemotherapy for advanced
non-small-cell lung cancer:results of a multicenter phase II study. J Clin
Oncol 19 : 1750-1758, 2001.
183) Fujiwara T, Kataoka M, Kawamata O, Tanaka N. [A phase I trial of adenoviral
p53 gene therapy for non-small cell lung cancer]. Nippon Geka Gakkai Zasshi 100 :
749-755, 1999.
184) Shimada H, Matsubara H, Ochiai T. p53 gene therapy for esophageal cancer. J
Gastroenterol 37 : 87-91, 2002.
185) Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, Ng L, Nye JA,
Sampson-Johannes A, Fattaey A, McCormick F. An adenovirus mutant that replicates
selectively in p53-deficient human tumor cells. Science 274 : 373-376,
1996.
186) Hecht JR, Bedford R, Abbruzzese JL, Lahoti S, Reid TR, Soetikno RM, Kirn
DH, Freeman SM. A phase I/II trial of intratumoral endoscopic ultrasound injection
of ONYX-015 with intravenous gemcitabine in unresectable pancreatic carcinoma. Clin
Cancer Res 9 : 555-561, 2003.
187) Hamid O, Varterasian ML, Wadler S, Hecht JR, Benson A, 3rd, Galanis E,
Uprichard M, Omer C, Bycott P, Hackman RC, Shields AF. Phase II trial of intravenous
CI-1042 in patients with metastatic colorectal cancer. J Clin Oncol 21 :
1498-1504, 2003.
188) Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein
B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362 :
857-860, 1993.
189) Lin J, Chen J, Elenbaas B, Levine AJ. Several hydrophobic amino acids
in the p53 amino-terminal domain are required for transcriptional activation,
binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev 8 :
1235-1246, 1994.
190) Roth J, Dobbelstein M, Freedman DA, Shenk T, Levine AJ. Nucleo-cytoplasmic
shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via
a pathway used by the human immunodeficiency virus rev protein. Embo J 17 :
554-564, 1998.
191) Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich
NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation
domain. Science 274 : 948-953, 1996.
192) Majeux N, Scarsi M, Caflisch A. Efficient electrostatic solvation model
for protein-fragment docking. Proteins 42 : 256-268, 2001.
193) Duncan SJ, Gruschow S, Williams DH, McNicholas C, Purewal R, Hajek M,
Gerlitz M, Martin S, Wrigley SK, Moore M. Isolation and structure elucidation
of Chlorofusin, a novel p53-MDM2 antagonist from a Fusarium sp. J Am Chem Soc 123 :
554-560, 2001.
194) Picksley SM, Vojtesek B, Sparks A, Lane DP. Immunochemical analysis of
the interaction of p53 with MDM2;─ fine mapping of the MDM2 binding site on
p53 using synthetic peptides. Oncogene 9 : 2523-2529, 1994.
195) Garcia-Echeverria C, Chene P, Blommers MJ, Furet P. Discovery of potent
antagonists of the interaction between human double minute 2 and tumor suppressor
p53. J Med Chem 43 : 3205-3208, 2000.
196) Chene P, Fuchs J, Carena I, Furet P, Garcia-Echeverria C. Study of the
cytotoxic effect of a peptidic inhibitor of the p53-hdm2 interaction in tumor
cells. FEBS Lett 529 : 293-297, 2002.
197) Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong
N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of
the p53 pathway by small-molecule antagonists of MDM2. Science 303 :
844-848. Epub 2004 Jan 2002, 2004.
198) Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of
mutant p53 conformation and function. Science 286 : 2507-2510,
1999.
199) Wang W, Takimoto R, Rastinejad F, El-Deiry WS. Stabilization of p53 by
CP-31398 inhibits ubiquitination without altering phosphorylation at serine
200) Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P,
Bergman J, Wiman KG, Selivanova G. Restoration of the tumor suppressor function
to mutant p53 by a low-molecular-weight compound. Nat Med 8 : 282-288,
2002.
201) Peng Y, Li C, Chen L, Sebti S, Chen J. Rescue of mutant p53 transcription
function by ellipticine. Oncogene 22 : 4478-4487, 2003.
202) Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov
MV, Gudkov AV. A chemical inhibitor of p53 that protects mice from the side
effects of cancer therapy. Science 285 : 1733-1737, 1999.
203) Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to
radiotherapy. Nat Rev Cancer 3: 117-129, 2003.
204) Komarova EA, Gudkov AV. Chemoprotection from p53-dependent apoptosis:potential
clinical applications of the p53 inhibitors. Biochem Pharmacol 62 :
657-667, 2001.
p53 and human cancer
Chikashi ISHIOKA
Department of Clinical Oncology, Institute of Development, Aging and Cancer,
Tohoku University and Tohoku University Hospital
Mutations in the TP53 gene are most frequent genetic alteration in human cancer, establishing the status of p53 protein as the central position in tumor suppressive machinery. Much has been learned during the past two decades about how p53 contributes to maintain genetic and cellular integrity against varied types of stresses, and also how disruption of p53 pathway involves in cancer progression, chemo-radio sensitivity and prognosis. Although incomplete, our current understanding of TP53 mutation illustrates how diverse mutations can affect molecular network in the p53 pathway. Understanding the p53 pathway and the inactivation mode of the pathway in more detail will provide insights into molecular mechanisms of tumor progression, chemo-radio sensitivity, and prognosis of disease, and will provide strategies to identify novel molecular target for cancer treatment.
Key words: p53, tumor suppressor gene, cancer, mutation
Kareiigaku Kenkyusho Zasshi 56(1), 1-34, 2004.